クリロフ量子対角化
このクリロフ量子対角化(KQD)のレッスンでは、以下の問いに答えます:
- クリロフ法とは一般的に何か?
- クリロフ法はなぜ機能するのか、またどのような条件下で機能するのか?
- 量子コンピューティングはどのような役割を果たすのか?
計算の量子部分は主に文献 [1] の研究に基づいています。
以下の動画では、古典コンピューティングにおけるクリロフ法の概要、その有用性の動機、そして量子コンピューティングがそのワークストリームにどのような役割を果たせるかを説明しています。続くテキストではより詳細な説明と、古典的手法および量子コンピュータを使用したクリロフ法の実装を提供します。
1. クリロフ法の概要
__クリロフ部分空間法__とは、__クリロフ部分空間__と呼ばれるものを基盤とした、いくつかの手法の総称です。これらの完全な解説はこのレッスンの範囲を超えますが、文献 [2-4] にはより多くの背景情報が記載されています。ここでは、クリロフ部分空間とは何か、固有値問題の解法においてなぜ有用であるか、そして最終的にそれを量子コンピュータ上でどのように実装できるかに焦点を当てます。
定義: 対称かつ半正定値の 行列 が与えられたとき、次数 のクリロフ空間 は、参照ベクトル に行列 の高次べき乗( まで)を掛けることで得られるベクトルによって張られる空間です。
上記のベクトルはクリロフ部分空間を張りますが、それらが直交しているとは限りません。そのため、__グラム・シュミット直交化__に似た反復的な正規直交化プロセスをよく使用します。ここでのプロセスは若干異なり、各新しいベクトルは生成される際に他のベクトルと直交するようにされます。この文脈ではこれを__アルノルディ反復__と呼びます。初期ベクトル から始め、次のベクトル を生成し、この2番目のベクトルが最初のベクトルと直交するように、 への射影を引き算します。すなわち
次のことから であることは容易に確認できます:
次のベクトルについても同様に、前の2つのベクトルの両方と直交するようにします:
このプロセスを 個すべてのベクトルについて繰り返すと、クリロフ空間の完全な正規直交基底が得られます。ここでの直交化プロセスは になると零ベクトルを生じることに注意してください。これは 個の直交ベクトルが必然的に全空間を張るためです。また、任意のベクトルが の固有ベクトルである場合、後続のすべてのベクトルがそのベクトルの定数倍となるため、プロセスも零ベクトルを生じます。
1.1 簡単な例:手計算によるクリロフ法
クリロフ部分空間の生成プロセスを、非常に小さな行列でステップごとに確認してみましょう。対象となる初期行列 から始めます:
この小さな例では、固有ベクトルと固有値を手計算でも求められます。ここでは数値解を示します。
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
後で比較するためにここに記録しておきます:
このプロセスがクリロフ部分空間の次元 を増やすにつれてどのように機能する(あるいは失敗する)かを調べたいと思います。そのために、以下のプロセスを適用します:
- ランダムに選んだベクトル (すでに正規化されている場合は と呼ぶ)から始め、全ベクトル空間の部分空間を生成する。
- 全行列 をその部分空間に射影し、射影された行列 の固有値を求める。
- グラム・シュミット直交化に似たプロセスを使用して、より多くのベクトルを生成し(正規直交性を確保しながら)、部分空間のサイズを増やす。
- をより大きな部分空間に射影し、結果として得られる行列 の固有値を求める。
- 固有値が収束するまで(このトイの例では元の行列 の全ベクトル空間を張るベクトルを生成するまで)これを繰り返す。
クリロフ法の通常の実装では、構築中のすべてのクリロフ部分空間について射影行列の固有値問題を解く必要はありません。目的の次元の部分空間を構築し、その部分空間に行列を射影してから、射影行列を対角化することができます。各部分空間次元での射影と対角化は、収束を確認するためにのみ行われます。
次元 :
ランダムなベクトル、例えば
を選びます。
正規化されていない場合は正規化します。
次に、行列 をこの1つのベクトルの部分空間に射影します:
これが という1つのベクトルのみを含むクリロフ部分空間への行列の射影です。この行列の固有値は自明に4です。これを の固有値(この場合は1つのみ)の0次近似と考えることができます。精度は低いですが、正しい桁数のオーダーです。
次元 :
次に、前のベクトルに を作用させて部分空間の次のベクトルを生成します:
次に、直交性を確保するために、このベクトルから前のベクトルへの射影を引き算します。
正規化されていない場合は正規化します。この場合、ベクトルはすでに正規化されているので、
次に、これら2つのベクトルの部分空間に行列 A を射影します:
この行列の固有値を求めるという問題が依然として残りますが、この行列は元の行列よりも少し小さくなっています。非常に大きな行列を扱う問題では、このより小さな部分空間を使用することが大きな利点となり得ます。
これはまだ良い近似ではありませんが、0次近似よりは改善されています。プロセスを明確にするためにもう1回繰り返します。ただし、次の反復では3×3行列を対角化することになるため、時間や計算能力の節約にはならないという点で、この手法の本来の利点を損なっています。
次元 :
次に、前のベクトルに A を作用させて部分空間の次のベクトルを生成します:
次に、直交性を確保するために、このベクトルから前の2つのベクトルへの射影を引き算します。
正規化されていない場合は正規化します。この場合、ベクトルはすでに正規化されているので、
次に、これらのベクトルの部分空間に行列 を射影します:
固有値を求めます:
これらの固有値は元の行列 の固有値と完全に一致しています。クリロフ部分空間を元の行列 の全ベクトル空間を張るように拡張したので、これは必然的な結果です。
この例では、クリロフ法が直接対角化よりも特に簡単に見えないかもしれません。実際、後のセクションで見るように、クリロフ法は特定の行列次元を超えた場合にのみ有利であり、これは非常に大きな行列の固有値・固有ベクトル問題を解くためのものです。
これが「手計算」で示す唯一の例ですが、以下のセクション2では計算機を使った例を示します。
用語の明確化
よくある誤解として、与えられた問題に対してクリロフ部分空間はただ1つしか存在しないというものがあります。しかし当然のことながら、行列を適用できる初期ベクトルは多数あるため、可能なクリロフ部分空間も多数存在します。「__その__クリロフ部分空間」という表現は、特定の例ですでに定義された特定のクリロフ部分空間を指す場合にのみ使用します。一般的な問題解決アプローチについては、「__ある__クリロフ部分空間」と呼びます。
最後の明確化として、「クリロフ__空間__」と呼ぶことも有効です。初期空間から部分空間へ行列を射影するという文脈で使用されることが多いため、「クリロフ__部分空間__」と呼ばれることが多いです。その文脈に合わせて、ここでは主に部分空間と呼びます。
理解度チェック
クリロフ部分空間の次元 を、対象となる行列の次元 を超えて拡張することが(a)有用でなく、(b)不可能である理由を説明してください。
Answer
(a) ベクトルを生成しながら正規直交化しているため、 個のそのようなベクトルは完全な基底を形成します。つまり、それらの線形結合を使用して空間内の任意のベクトルを作ることができます。
(b) 直交化プロセスは、新しいベクトルからすべての前のベクトルへの射影を引き算することで構成されます。前のベクトルが全ベクトル空間を張っている場合、全部分空間への射影を引き算すると常に零ベクトルが残ります。
同僚の研究者がクリロフ法を小さなトイ行列に適用して示そうとしています。行列 と初期ベクトル の選択に問題はありますか?
および
Answer
同僚は初期ベクトルとして偶然に固有ベクトルを選んでしまいました。初期ベクトルに行列を作用させると、固有値でスケールされた同じベクトルが返ってくるだけです。これでは次元が増加する部分空間を生成できません。同僚に別の初期ベクトルを選ぶよう、固有ベクトルでないものを確認してからアドバイスしてください。
適切な新しい初期ベクトルを選択して、指定の行列にクリロフ法を適用してください。クリロフ部分空間の0次と1次における最小固有値の推定値を書き下してください。
Answer
初期ベクトルの選択によって多くの可能な答えがあります。ここでは次を選びます:
を得るために、 を に1回作用させ、その後 を と直交させます。
0次では、クリロフ部分空間への射影は
1次では、このクリロフ部分空間への射影は
これは手計算でもできますが、numpyを使うのが最も簡単です:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
出力:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
最小固有値の推定値は -0.414 です。
1.2 クリロフ法の種類
「クリロフ部分空間法」とは、大規模な線形方程式系と固有値問題を解くために用いられるいくつかの反復手法の総称です。これらすべてに共通しているのは、クリロフ部分空間から近似解を構築するという点です。
ここで は初期推定値です(参考文献 [5] 参照)。各手法は、収束速度・メモリ使用量・全体的な計算コストのバランスを取りながら、この部分空間から最良の近似を選ぶ方法が異なります。このレッスンの焦点は、クリロフ部分空間法の文脈で量子計算を活用することです。これらの手法を網羅的に論じることはスコープ外となります。以下の簡潔な定義はあくまで参考のためであり、さらに詳しく調べるためのいくつかの参考文献を含んでいます。
共役勾配(CG)法: この手法は、対称正定値の線形方程式系を解くために使われます[6]。各反復において誤差の -ノルムを最小化するため、離散化された楕円型偏微分方程式から生じる方程式系に特に有効です[7]。次のセクションでは、線形方程式系のより良い解を探す上でなぜクリロフ部分空間が有効な部分空間となるのかを説明するために、このアプローチを活用します。
一般化最小残差(GMRES)法: 一般的な非対称線形方程式系を解くために設計された手法です。各反復においてクリロフ空間上で残差ノルムを最小化するため堅牢ですが、大規模な方程式系ではメモリ消費が大きくなる可能性があります[7]。
最小残差(MINRES)法: 対称不定値の線形方程式系を解くために使われます。GMRESと似ていますが、行列の対称性を利用して計算コストを削減します[8]。
その他の注目すべきアプローチとして、固有値問題に対するアーノルディ法と密接に関連した 完全直交化法(FOM)、双共役勾配(BiCG)法、誘導次元削減(IDR)法 があります。
1.3 クリロフ部分空間法がうまく機能する理由
ここでは、最急降下法の観点から、クリロフ部分空間法が反復的な固有ベクトル近似の改善を通じて行列固有値を効率的に近似できる理由を示します。基底状態の初期推定値が与えられた場合、その初期推定値に対して最も速い収束をもたらす逐次的な補正のなす空間がクリロフ部分空間であることを論じます。収束挙動の厳密な証明は行いません。
注目する行列 が対称正定値であると仮定します。これにより、以下の議論は上記のCG法に最もよく対応します。ここでは疎性について仮定は設けません。また、 がエルミート行列でなければならないとも主張しません(ハミルトニアンである場合は必要ですが)。
通常、次の形の問題を解くことを目的とします。
固有値問題のように ( はある定数)と考えることもできますが、ここでは問題設定をより一般的なまま保ちます。
近似解として ベクトル から始めます。この推定値 とセクション1.1の との間には類似点がありますが、ここではそれを活用しません。推定値 には誤差があり、それを と呼びます:
また残差 を次のように定義します:
ここでは、クリロフ部分空間の次元 と区別するため、大文字の を使います。

次に、次の形の補正ステップを行います。
これにより近似が改善されることが期待されます。ここで はまだ未定のベクトルです。補正後の誤差を とすると、

行列によって変換された後の誤差の挙動に関心があります。そこで、誤差の -ノルムを計算しましょう。すなわち
ここで の対称性と を利用しました。 は に依存しない定数です。セクション1.2で述べたように、誤差の -ノルムは最小化する唯一の量ではありませんが、適切な選択です。補正ベクトル の選び方に応じてこの量がどのように変化するかを見ていきます。そこで関数 を次のように定義します。
は補正 の関数として -ノルムで測った誤差 にほかなりません。したがって、 を最小化するように を選びたいのです。この目的のため、 の勾配を計算します。 の対称性を使うと
勾配は最急上昇の方向を指しているので、その反対方向が関数が最も大きく減少する方向、すなわち 最急降下 の方向となります。初期推定値 (すなわち )において、 つまり、関数 は残差 の方向に最も大きく減少します。そのため、初期選択としては ( はスカラー)を加えることが最も有益です。
次のステップでは、再びベクトル を選び、現在の近似値に加えます。先ほどと同じ議論から、( はスカラー)を選びます。この操作を繰り返すと、 番目の反復でのベクトルは
同等に言えば、改善された推定値を選ぶ空間を 、、... の順に加えていくことで構築します。 番目の推定ベクトルは次の空間に属します。
次の関係を使うと、
次のことがわかります。
つまり、正しい解 を最も効率的に近似するために構築される空間は、まさに行列 を に逐次的に作用させることで構築される空間です。クリロフ部分空間は、最急降下の方向の逐次的なベクトルが張る空間にほかなりません。
最後に、このアプローチのスケーリングに関する数値的な主張は行っておらず、疎行列に対する比較優位についても論じていないことを改めて強調しておきます。これはあくまでクリロフ部分空間法の活用を動機付け、直感的な理解を与えることを目的としています。次はこれらの手法の数値的な挙動を探っていきます。
理解度の確認
上記のワークフローでは、誤差の -ノルムを最小化することを提案しました。基底状態とその固有値を求めるにあたって、他にどのような量を最小化することが考えられるでしょうか?
Answer
誤差の -ノルムの代わりに残差ベクトルを使うことが考えられます。誤差ベクトルそのものを考慮することが有用なケースもあるかもしれません。
2. 古典計算におけるクリロフ法
このセクションでは、固有値問題を解くためにクリロフ部分空間を活用できるよう、アーノルディ反復を計算的に実装します。まず小規模な例に適用し、次に行列のサイズが増大するにつれて計算時間がどのようにスケールするかを検討します。ここで重要なアイデアは、クリロフ空間を張るベクトルの生成が、必要な合計計算時間の大きな部分を占めるということです。必要なメモリは特定のクリロフ法によって異なります。しかし、メモリの制約が従来のクリロフ法の利用を制限することがあります。
2.1 シンプルな小規模の例
クリロフ部分空間を構築する過程で、部分空間内のベクトルを正規直交化する必要があります。部分空間内の既存ベクトル vknown(正規化されているとは限らない)と、部分空間に追加する候補ベクトル vnext を受け取り、vnext を vknown と直交させかつ正規化する関数を定義しましょう。さらに、クリロフ部分空間内のすべての既存ベクトルに対してこの処理を順に行い、完全な正規直交集合を確保する関数を定義しましょう。
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
次に、元の行列の全空間をクリロフベクトルが張るまで、クリロフ部分空間を反復的に拡大していく関数を定義しましょう。これにより、クリロフ部分空間法で得られる固有値が正確な値とどの程度一致するかを、クリロフ部分空間の次元の関数として確認できます。重要な点として、関数 krylov_full_build はクリロフベクトル、射影ハミルトニアン、固有値、および所要時間を返します。
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
手で計算するには大きすぎますが、まだ比較的小さな行列でこれをテストしてみましょう。
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
最後のステップ(クリロフ空間が元の行列の全ベクトル空間となるとき)において、クリロフ法による固有値が厳密な数値対角化の値と完全に一致することを確認することで、関数の正しさを検証できます。
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
成功です。もちろん実際に重要なのは、クリロフ部分空間の次元の関数として近似がどれほど良いかです。基底状態やその他の最小固有値を求めることが多い(また後述するより代数的な理由から)ため、クリロフ部分空間の次元の関数として最小固有値の推定値を見てみましょう。
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
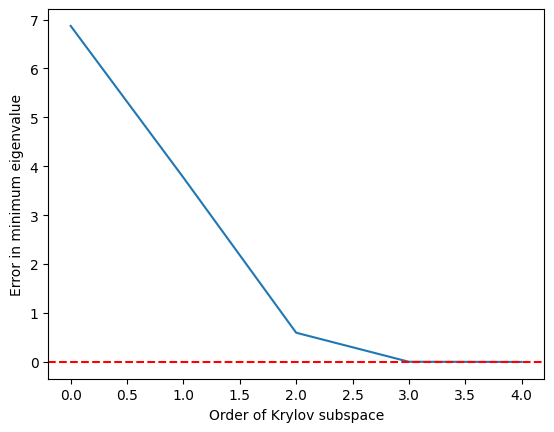
クリロフ部分空間が まで大きくなるとかなり正確に最小固有値に到達し、 では完全に一致することがわかります。
2.2 行列次元に対する時間スケーリング
次の方法で、クリロフ法が厳密な数値固有値ソルバーより有利になり得ることを確認しましょう。
- ランダム行列を構築する(疎でなく、KQDの理想的な用途ではない)
- 2つの方法で固有値を求める:NumPy を使った直接法と、クリロフ部分空間を使った方法
- クリロフによる推定値を受け入れる前に必要な精度の閾値を設定する
- 2つの方法に要する実時間を比較する
注意点: 後で詳しく説明するように、クリロフ量子対角化(KQD)は、行列表現が疎であるか、少数の交換可能なパウリ演算子のグループで書ける演算子に最も有効に適用されます。ここで使用するランダム行列はその条件を満たしていません。これらはあくまで、古典的なクリロフ法が有用になりうる規模を調べるためのものです。 次に、クリロフ法を使う際には、さまざまなサイズのクリロフ部分空間で固有値を段階的に計算します。基底状態固有値について必要な精度を達成した最小次元の部分空間の計算時間を記録します。これも、厳密な固有値ソルバーでは手に負えない問題を解くこととは少々異なります。なぜなら、必要な次元を評価するために厳密解を使っているからです。
まずランダム行列の集合を生成します。
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
次に、各行列を NumPy を使って直接対角化します。後の比較のため、対角化に要した時間を記録します。
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
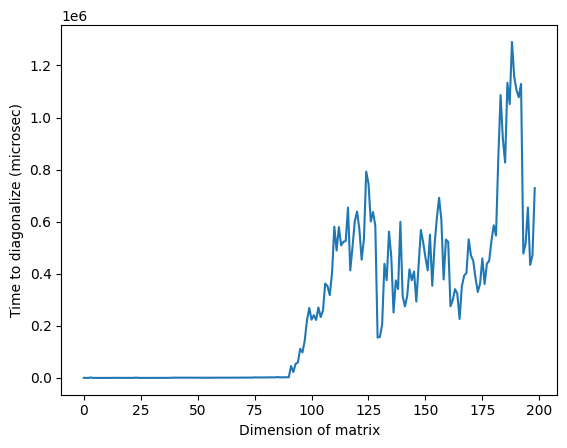
上の図において、次元が約125付近で異常に高い時間が見られますが、これは行列のランダム性やクラシックプロセッサの実装によるものであり、再現性はありません。コードを再実行すると、異なるプロファイルと異なる異常なピークが現れます。
次に、各行列に対してクリロフ部分空間を段階的に構築し、各ステップで固有値を計算します。各ステップで、最小固有値が指定した絶対誤差以内に得られているかどうかを確認します。指定した誤差以内の固有値を初めて与える部分空間について、計算時間を記録します。このセルの実行はプロセッサの速度によっては数分かかる場合があります。実行をスキップしたり、対角化する行列の最大次元を減らしたりして構いません。事前に計算された結果を見るだけでも十分です。
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
2つの方法で得られた時間を比較するためにプロットしましょう。
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

これらは実際に要した時間ですが、議論のために隣接する数点・行列次元の平均を取ることでこれらの曲線を平滑化してみましょう。以下でその処理を行います。
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

クリロフ部分空間の構築に要する時間は、当初は NumPy による完全対角化に要する時間を上回ります。しかし行列のサイズが大きくなるにつれて、クリロフ法が有利になります。これは許容誤差を小さくしても当てはまりますが、その場合は優位性が現れるのがより大きな行列サイズになります。この点を掘り下げてみましょう。
数値対角化の時間複雑度は (アルゴリズムによって多少の違いあり)です。 個のベクトルからなる正規直交基底を生成する時間複雑度も です。したがって、クリロフ法の優位性は 正規直交基底を使うことではなく、注目する固有値を効果的に取り出せる特定の正規直交基底を使うことに関係しています。このレッスンの第1節のスケッチ証明から既にわかっていたことであり、クリロフ法の収束保証にとって極めて重要です。
これまでの進捗を振り返りましょう。
- 非常に大規模な行列に対して、クリロフ部分空間法は従来の対角化アルゴリズムよりも速く、必要な許容誤差内の近似固有値を求められる可能性があります。
- そのような非常に大規模な行列では、クリロフ部分空間の生成がクリロフ部分空間法の中で最も時間のかかる部分です。
- したがって、クリロフ部分空間を効率的に生成する方法は非常に価値があります。
ここでついに量子コンピュータが登場します。
理解度チェック
上に示した対角化時間 vs. 行列次元の平滑化プロットを参照してください。
(a) このプロットによると、クリロフ法が速くなったのはおよそどの行列次元からですか?
(b) クリロフ法が速くなる次元を変えうる計算上の要因にはどのようなものがありますか?
Answer
(a) 計算を再実行すると答えは変わりますが、クリロフ法はおおよそ次元80〜85から速くなります。
(b) 考えられる答えは多数あります。重要な要因として、要求する精度や対角化する行列のスパース性などが挙げられます。
3. 時間発展によるクリロフ法
ここまで説明してきた内容はすべて古典的に実行できます。では、量子コンピューターをどのように・どのような場面で利用するのでしょうか?非常に大きな行列に対しては、クリロフ法は長い計算時間と大量のメモリを必要とする場合があります。 を に作用させる行列演算に要する時間は、最悪の場合 のスケーリングとなります。スパース行列をベクトルに乗じる場合(古典クリロフ型ソルバーでの典型的なケース)でも、時間複雑度は のスケーリングです。これを、部分空間に含めたいすべてのベクトルに対して行います。部分空間の次元 は通常 に対して有意な割合を占めず、多くの場合 程度のスケーリングです。したがって、すべてのベクトルの生成は最悪の場合 のスケーリングとなります。直交化など他のステップも存在しますが、このスケーリングが支配的な点として念頭に置いておく必要があります。
量子コンピューティングにより、問題のどの属性が時間・リソースのスケーリングを決定するかを変えることができます。行列サイズ への全面的な依存ではなく、ショット数やハミルトニアンを構成する非可換パウリ項の数といった要素が関わってきます。この仕組みを詳しく見ていきましょう。
3.1 時間発展
量子状態を時間発展させる演算子は であることを思い出してください(量子コンピューティングでは特に、表記から を省略することが一般的です)。演算子のこのような指数関数を理解し実現する一つの方法は、テイラー展開を見ることです。この演算を何らかの初期ベクトル に作用させると、 を初期状態に繰り返し作用させた項の和が得られます。初期推定状態を時間発展させるだけでクリロフ部分空間を構成できるように見えます!
注意点は、実際の量子コンピューターで時間発展を実現することにあります。ハミルトニアン中の多くの項は互いに非可換です。 のような単純な指数演算子は単純な回路に対応しますが、一般的なハミルトニアンはそうではありません。また、非可換な項を含むため、数の場合のように指数を単純な積に分解することはできません。
これは自明ではありませんが、量子コンピューティングにおいてよく研究されたプロセスです。量子コンピューター上での時間発展は、トロッタリゼーションと呼ばれる手法で実行します。これ自体が奥深いテーマです[10]。高いレベルで説明すると、時間発展を非常に小さなステップ、たとえば 個の ステップに分割することで、項の非可換性の影響を抑えることができます。
ここで です。
古典的な文脈で の冪乗を直接用いて生成した次数 のクリロフ部分空間を「べき乗クリロフ部分空間」と呼ぶことにします。
次に、ユニタリ時間発展演算子 を用いて同様の空間を生成します。これを「ユニタリクリロフ空間」 と呼びます。古典的に用いるべき乗クリロフ部分空間 は、 がユニタリ演算子でないため量子コンピューター上で直接生成することはできません。ユニタリクリロフ部分空間を用いても、べき乗クリロフ部分空間と同様の収束保証が得られることが示されています。すなわち、初期状態 が真の基底状態と指数的に消滅しない重なりを持ち、かつ固有値間に十分なギャップが存在する限り、基底状態誤差は効率的に収束します。収束についてのより厳密な議論は参考文献[1]を参照してください。
ここで、 の冪乗は異なる時刻ステップに対応します( の 乗は時刻 だけ前進させることに相当します)。合計時間 だけ時間発展させた部分空間の要素を と表します。
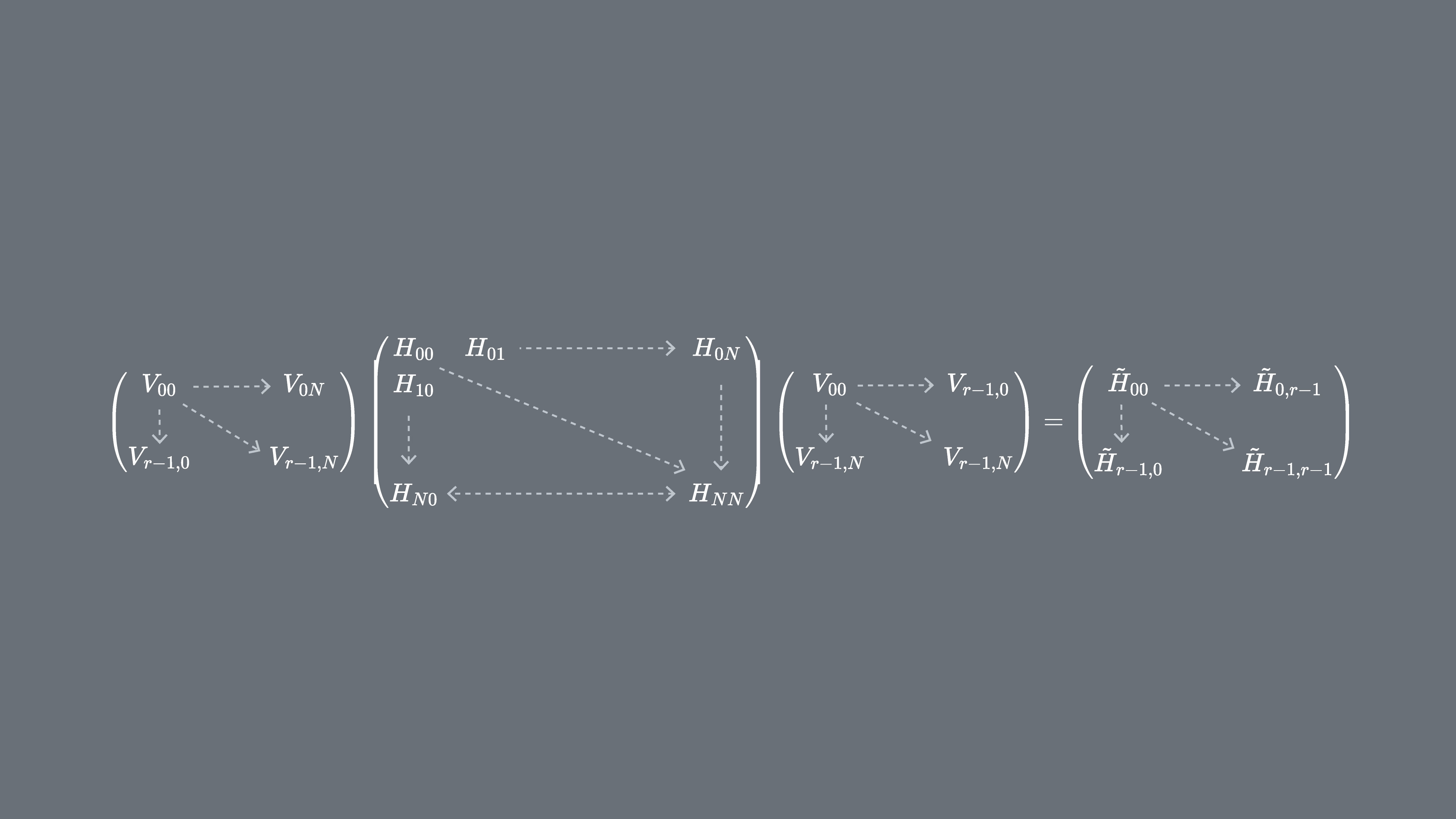
ハミルトニアン をユニタリクリロフ部分空間 に射影することができます。つまり、 基底における の各行列要素を計算します。この射影された行列を と呼びます。
3.2 量子コンピューターへの実装方法
の行列要素は期待値 で与えられ、これは量子コンピューターを使って推定できます。 はさまざまな量子ビット上のパウリ演算子の和として書けること、また、すべてのパウリ演算子を同時に測定することはできないことを念頭に置いてください。パウリ項を可換な項のグループに分類し、グループをまとめて測定することができます。しかし、すべての項をカバーするには多くのグループが必要になる場合があります。そのため、項を分割できる可換グループの数 が重要になります。
ここで は という形のパウリ列、または互いに可換なそのようなパウリ列の集合です。 をこのような測定可能な演算子の和として書けることを踏まえると、 の行列要素に関する以下の表式は Qiskit Runtime プリミティブの Estimator を使って実現できます。
ここで はユニタリクリロフ空間のベクトルであり、 は選択した時間ステップ の倍数です。量子コンピューター上での各行列要素の計算は、量子状態間の重なりを得るための任意のアルゴリズムで行うことができます。本レッスンではアダマールテストに焦点を当てます。 の次元が であるとき、部分空間に射影されたハミルトニアンは の次元を持ちます。 が十分に小さければ(一般に固有値の推定値が収束するには で十分です)、射影されたハミルトニアン を古典的に容易に対角化できます。ただし、クリロフ空間のベクトルは非直交であるため、 を直接対角化することはできません。それらの重なりを測定し、行列 を構築する必要があります。
これにより、非直交空間における固有値問題(一般化固有値問題とも呼ばれます)を解くことができます。
この一般化固有値問題の解を見ることで、 の固有値と固有状態の推定値が得られます。たとえば、基底状態エネルギーの推定値は最小の固有値 を取ることで得られ、基底状態は対応する固有ベクトル から得られます。 の係数が を張る異なるベクトルの寄与を決定します。
一般化固有値問題
なぜ を単純に対角化できないのでしょうか? はクリロフ基底の幾何学的な情報(ごく特殊なケースを除き非直交)を含んでいるため、 単体では全ハミルトニアンの射影を記述しておらず、その固有値は全ハミルトニアンの固有値と特別な関係を持ちません——どのような値にもなりえます。全ハミルトニアンのクリロフ空間への射影に対応する近似固有値・固有ベクトルを得るには、一般化固有値問題を解く必要があります。
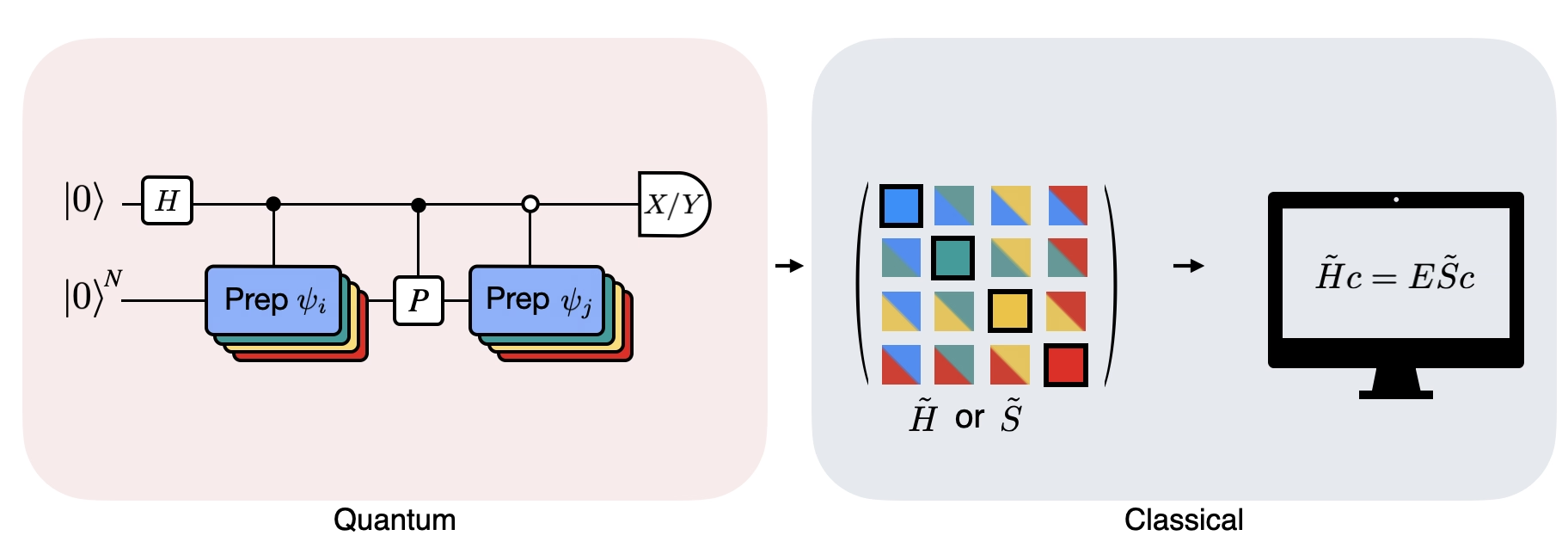
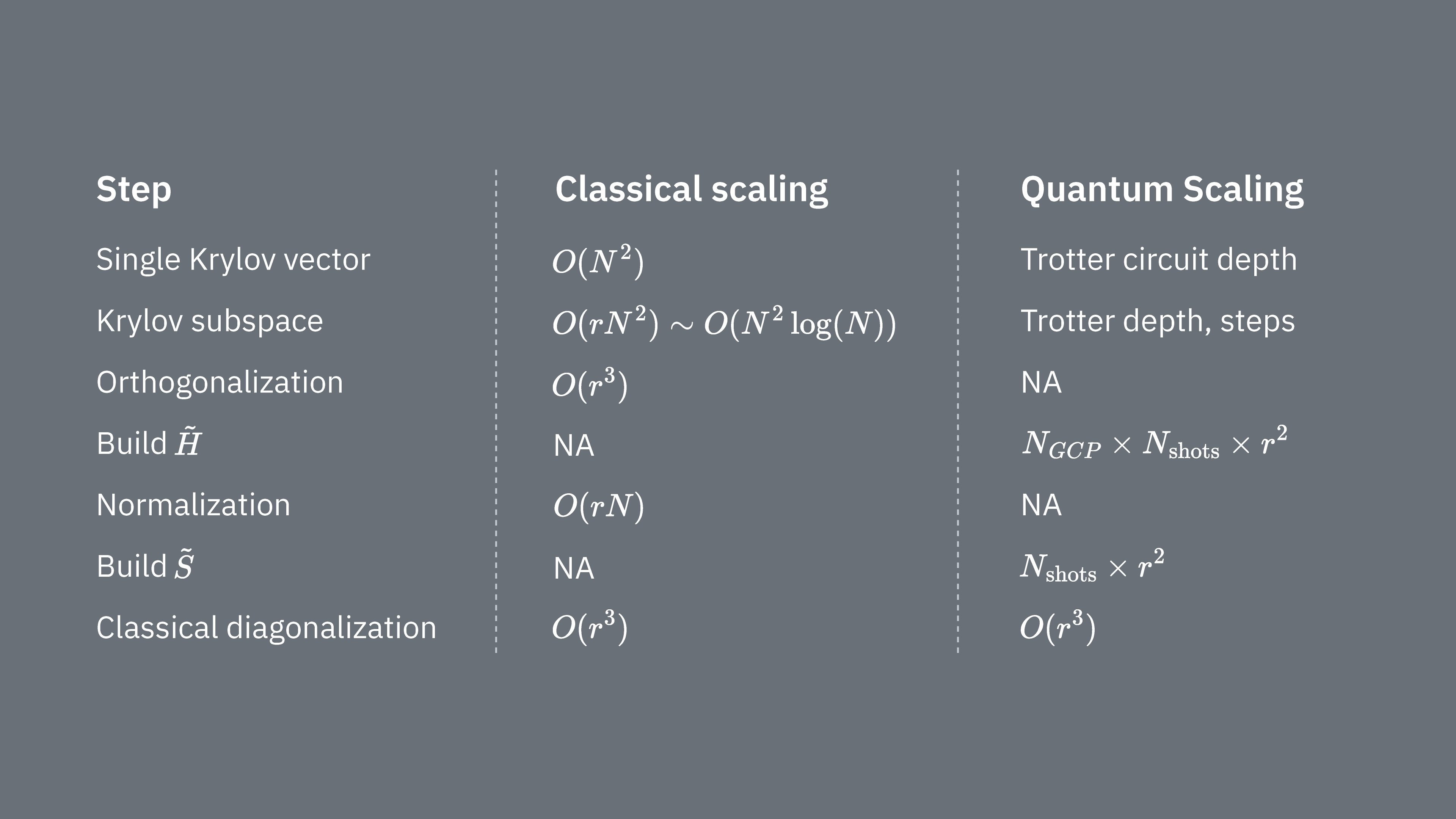
この図は、異なる量子状態間の重なりを計算するために用いられる修正アダマールテストの回路表現を示しています。各行列要素 に対して、状態 、 間のアダマールテストが実施されます。これは、行列要素の色分けと対応する 、 操作によって図中に強調されています。したがって、射影されたハミルトニアン のすべての行列要素を計算するには、クリロフ空間ベクトルのすべての組み合わせに対するアダマールテストのセットが必要です。アダマールテスト回路の最上部のワイヤは補助量子ビットであり、X 基底または Y 基底で測定されます。その期待値が状態間の重なりの値を決定します。下部のワイヤはシステムのハミルトニアンのすべての量子ビットを表します。 操作は、補助量子ビットの状態によって制御されながら、システム量子ビットを状態 に準備します( も同様)。演算 はシステムハミルトニアン のパウリ分解を表します。量子コンピューター上でのこの実装は、以下でより詳しく示します。
4. 量子コンピューター上でのクリロフ量子対角化
いよいよ実際の量子コンピューターでクリロフ量子対角化を実装します。まず、いくつかの便利なパッケージをインポートしましょう。
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
上で説明した一般化固有値問題を解くための関数を以下に定義します。
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
初期ベンチマーキングでは、収束挙動を確認するために厳密な古典解を知っておくことが有用です。以下の関数は、ハミルトニアンと量子ビット数を引数として、ハミルトニアンの基底状態エネルギーを計算します。
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 ステップ1:問題を量子回路と演算子にマッピングする
次にハミルトニアンを定義します。上の関数はハミルトニアンを引数として受け取り基底状態のみを古典的に返すものですが、ここで定義するハミルトニアンはすべてのエネルギー固有状態のエネルギー準位を決定するものであり、パウリ演算子を用いて構成し量子コンピューター上で実装できます。
ここでは空間内の任意の向きを持つスピンの鎖に対応するハミルトニアン「ハイゼンベルク鎖」を選択します。 番目のスピンは最近接隣接( 番目および 番目のスピン)のみに影響を受け、より遠い隣接には影響されないと仮定します。また、スピンが異なる軸方向を向く場合にスピン間の相互作用が異なる可能性も考慮します。この異方性は、たとえばスピンが埋め込まれた結晶格子の構造に起因して生じることがあります。
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
以下のコードはハミルトニアンを一粒子状態に制限し、スペクトルノルムを用いて適切な時間ステップ のサイズを設定します。時間ステップ dt の値をヒューリスティックに選択します(ハミルトニアンのノルムの上界に基づきます)。参考文献[9]は、十分に小さい時間ステップとして が適切であり、この値を過大評価するよりも過小評価する方が望ましいことを示しています。過大評価すると高エネルギー状態の寄与がクリロフ空間内の最適状態さえも損なう可能性があります。一方、 を小さくしすぎると、時間ステップごとにクリロフ基底ベクトルの差異が小さくなるため、クリロフ部分空間の条件数が悪化します。
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
時間発展に使用するトロッター・ステップ数を指定します。また、最大クリロフ次元を4と指定します。このクリロフ次元は現実的なアプリケーションには十分ではありませんが、この例では十分です。さらに、より小さな次元でも収束を確認します。後のレッスンでは、ハミルトニアンをより大きな部分空間にスケールアップして射影できる方法を探っていきます。
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
状態準備
基底状態と何らかの重なりを持つ参照状態 を選びます。このハミルトニアンでは、中央の量子ビットに励起を持つ状態 を参照状態として使用します。
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

時間発展
与えられたハミルトニアンが生成する時間発展演算子 は、Lie-Trotter近似を通じて実現できます。簡単のため、時間発展回路には組み込みの PauliEvolutionGate を使用します。一般的な構文は次のとおりです。
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
これをアダマールテストの中で、時刻 ずつ前進するかたちで以下のように使用します。
アダマールテスト
とグラム行列 の両方の行列要素をアダマールテストを使って計算したいことを思い出してください。この文脈でどのように機能するかを確認しましょう。まず の構成に焦点を当てます。全体的なプロセスを以下にグラフィカルに示します。色分けされた状態準備ブロック の複数の層は、このプロセスが部分空間内の と のすべての組み合わせに対して実行されることを思い起こさせます。
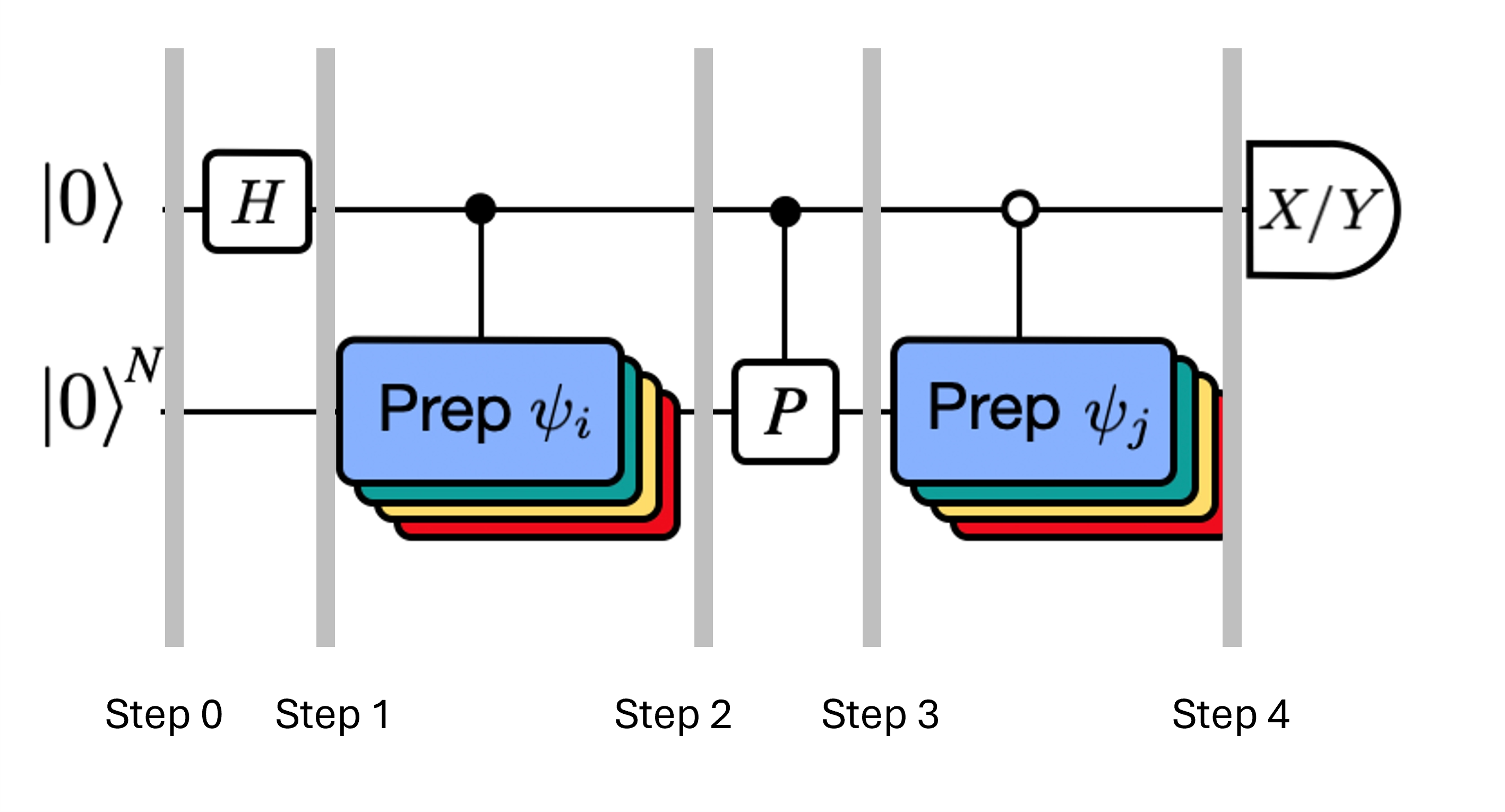
各ステップでのシステムの状態は次のとおりです。
ここで はハミルトニアンの分解におけるパウリ項です(複数の可換パウリ項の線形結合はユニタリでないため使用できないことに注意してください——グループ化は後で示す別の構成を使って可能です)。、 はユニタリクリロフ空間のベクトル 、 を準備する制御演算であり、 です。この回路に と の測定を適用することで、それぞれ必要な行列要素の実部と虚部が計算されます。
上のステップ4から始め、ゼロ番目の量子ビットにアダマールゲート を適用します。
次に または を測定します。
恒等式 より。同様に、 を測定すると次が得られます。
これらのステップを前に設定した時間発展に追加して、以下のように記述します。
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

トロッター回路の深さについては既に注意しました。この条件でアダマールテストを実行すると、特にネイティブゲートに分解すると、さらに深い回路になる可能性があります。デバイスのトポロジーを考慮すると、これはさらに増加します。したがって、量子コンピューターの時間を使う前に、回路の2量子ビットゲート深さを確認しておくことが重要です。
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
この深さの回路では、現代の量子コンピューターで使用可能な結果を得ることはできません。 と を構築するには、より優れた方法が必要です。これが以下で紹介する効率的なアダマールテストの背景にある理由です。
4.2 ステップ2:ターゲットハードウェア向けに回路と演算子を最適化する
効率的なアダマールテスト
いくつかの近似を導入し、モデルハミルトニアンに関するある仮定を利用することで、アダマールテスト用に得られた深い回路を最適化できます。例として、次のアダマールテスト回路を考えます:
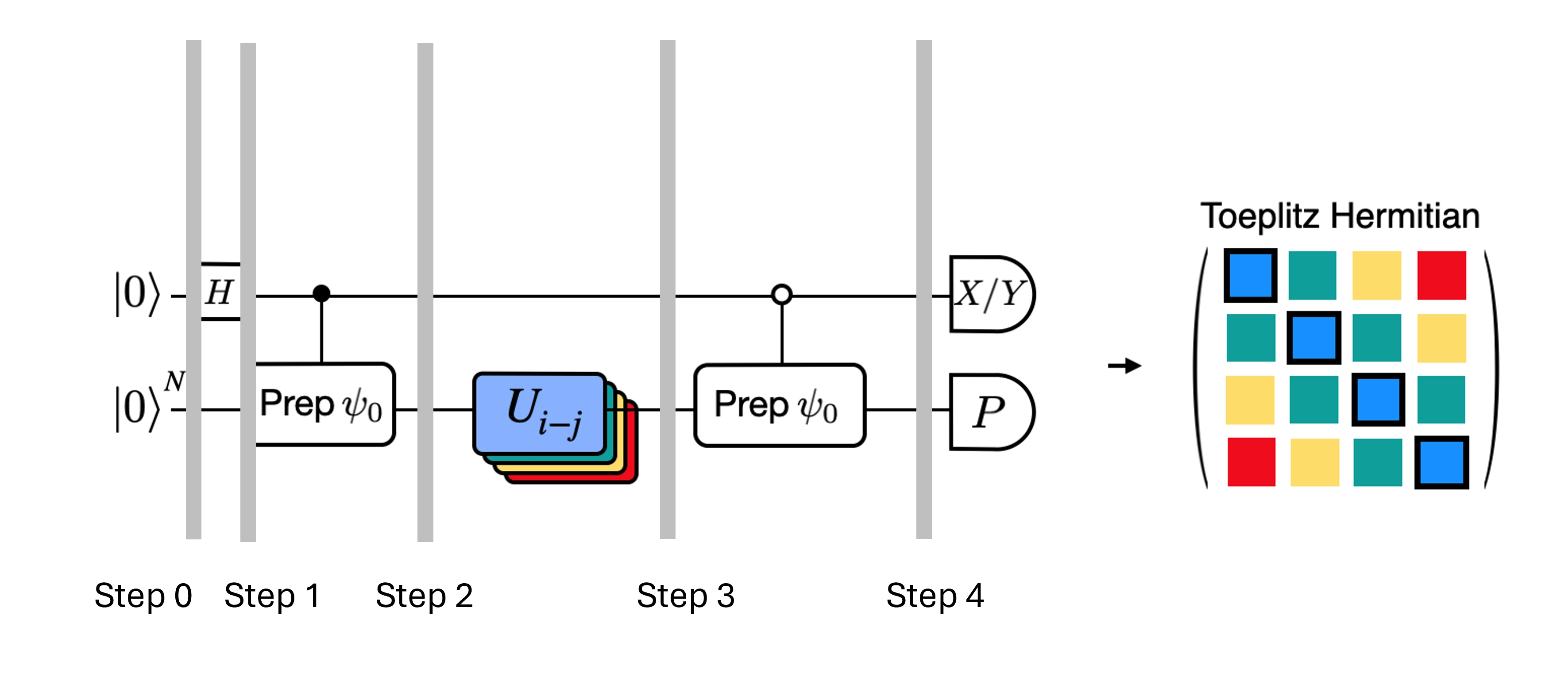
ハミルトニアン のもとで の固有値である を古典的に計算できると仮定します。 これは、ハミルトニアンが U(1) 対称性を保持する場合に満たされます。これは強い仮定のように見えるかもしれませんが、ハミルトニアンの作用を受けない真空状態(この場合は 状態に対応)が存在すると安全に仮定できる場合が多くあります。これは例えば、電子数が保存される安定分子を記述する化学ハミルトニアンに当てはまります。 ゲート が所望の参照状態 を準備するとすれば、例えば化学の HF 状態を準備するために は単一量子ビット NOT のテンソル積となり、制御付き は単純に CNOT のテンソル積になります。 このとき、上記の回路は測定前に次の状態を実装します:
ここで、ステップ 2 から 3 への古典的にシミュレート可能な位相シフト を使用しています。したがって期待値は次のようになります:
これらの仮定を用いることで、制御演算を減らして対象演算子の期待値を書き表すことができました。実際、制御付き状態準備 のみを実装すれば済み、制御付き時間発展は必要ありません。上記のように計算を再定式化することで、得られる回路の深さを大幅に削減できます。
なお、ボーナスとして、パウリ演算子が回路の途中の制御ゲートとしてではなく、回路の最後の測定として現れるようになるため、上記で与えた分解 のように、他の可換パウリ演算子と合わせて同時に測定できます。
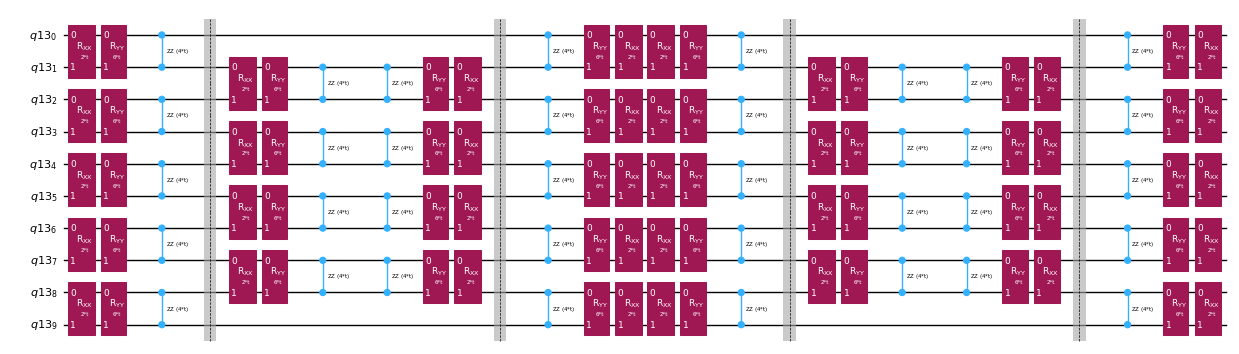
トロッター分解による時間発展演算子の分解
時間発展演算子を厳密に実装する代わりに、トロッター分解を使ってその近似を実装できます。特定の次数のトロッター分解を何度も繰り返すことで、近似から生じる誤差をさらに低減できます。以下では、考慮しているハミルトニアンの相互作用グラフ(最近接相互作用のみ)に対して最も効率的な方法でトロッター実装を直接構築します。具体的には、結合強度 、、 とパラメータ化された角度 を持つパウリ回転 、、 を挿入します。これは の近似実装に対応しています。パウリ回転の定義と実装しようとしている時間発展の定義の違いから、時間発展 を実現するにはパラメータ を使用する必要があります。さらに、奇数回のトロッターステップの繰り返しに対して演算の順序を逆にします。これは機能的には同等ですが、隣接する演算を単一の ユニタリに統合できるため、汎用の PauliEvolutionGate() 機能を使って得られる回路よりもはるかに浅い回路になります。
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
効率的なアダマールテストのために、再び初期状態を準備します。
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

アダマールテストによる と の行列要素計算のためのテンプレート回路
アダマールテストで使用される回路の唯一の違いは、時間発展演算子の位相と測定する観測量です。そのため、時間発展演算子に依存するゲートのプレースホルダーを持つ、アダマールテストの汎用回路を表すテンプレート回路を準備できます。
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
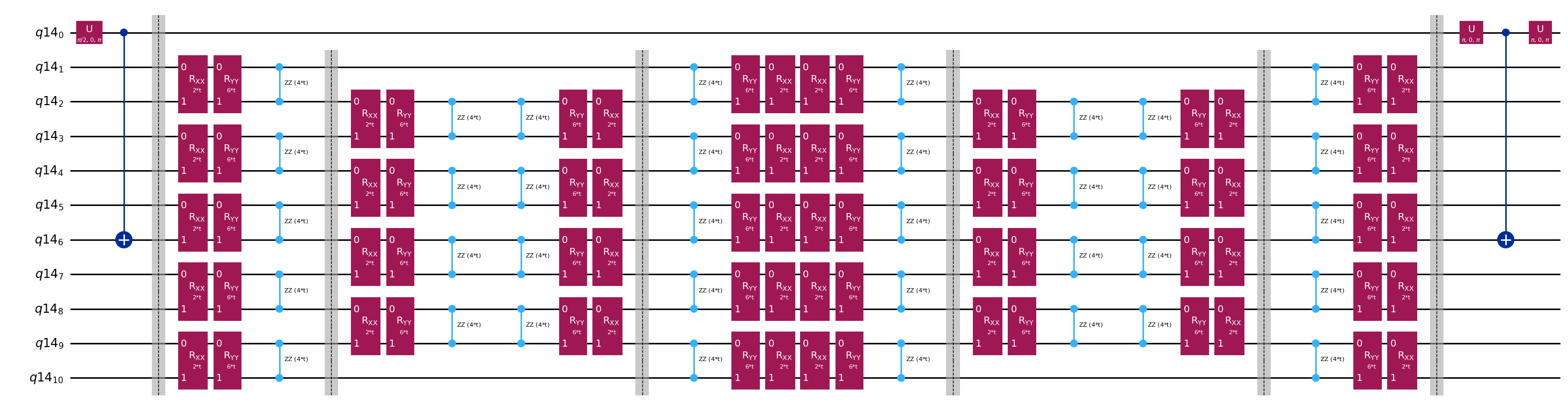
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
この深さは元のアダマールテストと比較して大幅に削減されています。この深さは現代の量子コンピュータで扱える範囲ですが、まだかなり高い値です。有用な結果を得るためには、最先端のエラー軽減技術を使用する必要があります。
量子クリロフ計算を実行するバックエンドを選択し、その量子コンピュータ上で実行するために回路をトランスパイルします。
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
次に、回路と演算子をトランスパイルします。
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

最適化後、トランスパイルされた2量子ビットゲートの深さがさらに削減されています。
4.3 ステップ3:Qiskit Runtime プリミティブを使用して実行する
Estimator で実行するための PUB を作成します。
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
の回路は古典的に計算可能です。量子コンピュータを使用する の場合に進む前に、この計算を実行します。
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
効率的なアダマールテストを使用することでゲートの深さを桁違いに削減できましたが、それでも最先端のエラー軽減技術が必要な深さです。以下では、使用する軽減手法の属性を指定します。すべての手法が重要ですが、特に確率的エラー増幅(PEA)について触れておく価値があります。この強力な手法は、多大な量子オーバーヘッドを伴います。ここで行う計算は、実際の量子コンピュータで実行するのに20分以上かかる場合があります。精度とオーバーヘッドを増減させるために、以下のパラメータを調整してみてください。以下のデフォルト設定は高品質な結果をもたらします。
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
最後に、Estimator を使って と の回路を実行します。
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 ステップ4:後処理と結果の分析
量子コンピュータから得られるのは、 の個々の行列要素と、 の行列要素を構成する可換パウリグループです。これらの項を組み合わせて行列を復元し、一般化固有値問題を解く必要があります。
# Store the outputs as 'results'.
results = job.result()[0]
有効ハミルトニアン行列とオーバーラップ行列の計算
まず、制御なしの時間発展の間に 状態が蓄積する位相を計算します。
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
回路実行の結果が得られたら、データを後処理して の行列要素を計算できます。
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
そして の行列要素を計算します。
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
最後に、 の一般化固有値問題を解きます:
そして基底状態エネルギー の推定値を得ます。
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
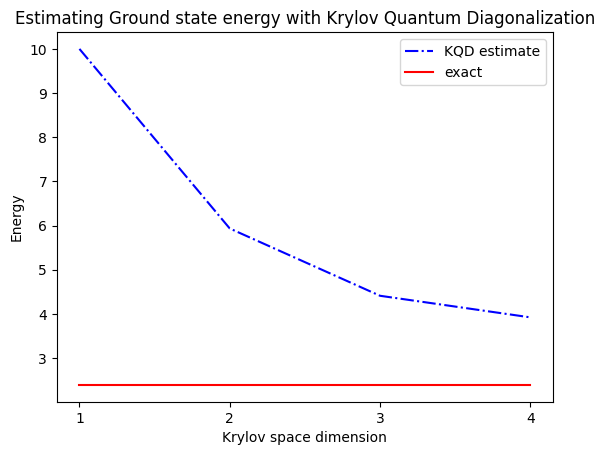
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
一粒子セクターの場合、このセクターのハミルトニアンの基底状態を古典的に効率よく計算できます。
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
5. 考察と発展
まとめると、まず参照状態から始め、異なる時間だけ発展させてユニタリ・クリロフ部分空間を生成します。次にハミルトニアンをその部分空間に射影します。また、部分空間ベクトルのオーバーラップを推定します。最後に、より低次元の一般化固有値問題を古典的に解きます。
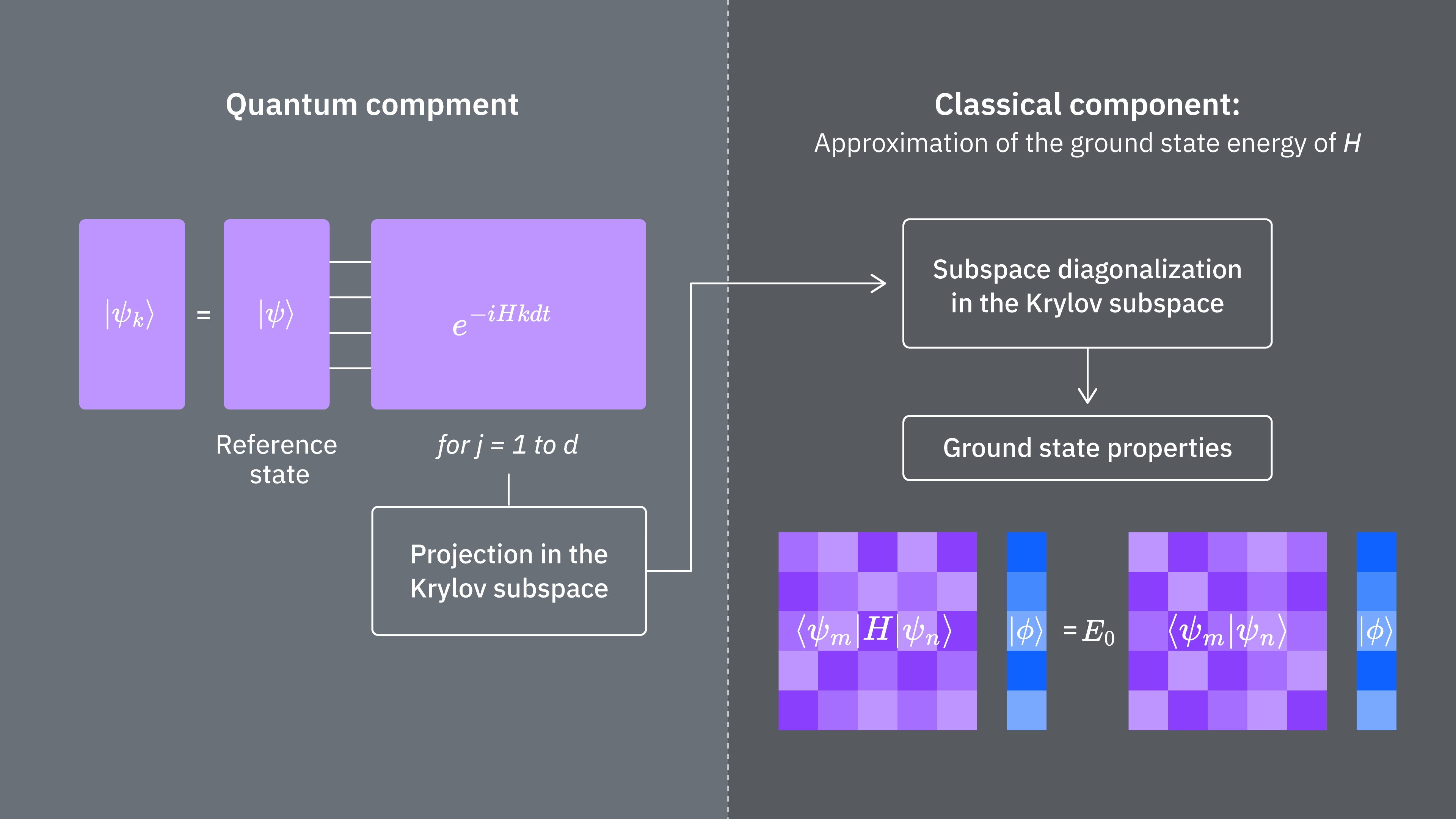
クリロフ法を古典的および量子力学的に使用した場合の計算コストを決定する要因を比較してみましょう。すべてのステップに完全に対応する古典・量子のアナロジーが存在するわけではありません。この表は、考慮すべき各ステップのスケーリングをまとめたものです。
ハミルトニアンには一般に同時測定できない(互いに非可換な)項が存在することを思い出してください。ハミルトニアンの項を、同時測定可能な可換パウリ演算子のグループに分類しますが、互いに非可換な全ての項を考慮するには多くのグループが必要になる場合があります。量子コンピュータ上で を構築するには、ハミルトニアン中の可換パウリ文字列の各グループに対して個別の測定が必要であり、それぞれ多数のショットを要します。これを 個の異なる行列要素に対して行う必要があり、異なる時間発展因子の 通りの組み合わせに対応します。これを削減する方法もありますが、この大まかな扱いでは、必要な時間は のようにスケールします。 の要素の推定には がかかります。最後に、射影空間における一般化固有値問題を古典的に解くには を要します。
量子クリロフ対角化は、ハミルトニアン中の可換パウリグループの数が比較的少ない場合に有用です。これらのスケーリング依存性は、クリロフ法が有効なアプリケーションとそうでないアプリケーションを示唆しています。 ハミルトニアンの中には、量子ビットへのマッピング時に高い複雑性を持ち、少数の可換グループに容易に分割できない多くの非可換パウリ文字列を含むものがあります。これは例えば量子化学の問題でよく当てはまります。この複雑性は、近期の量子コンピュータにとって2つの主要な課題をもたらします:
- の各要素の推定が、項の数が多いために計算コストが高くなります。
- 必要なトロッター回路が過度に深くなります。
上記の問題はいずれも、量子コンピュータがフォールトトレランスに達したときに少なくなりますが、近期においては考慮が必要です。量子化学よりも「単純な」マッピングを持つ系でさえ、ハミルトニアンに非可換項が多すぎる場合、同様の障害に直面する可能性があります。 クリロフ法は、ハミルトニアンが比較的少数の可換パウリグループに分割でき、 がトロッター回路で実装しやすい場合に最も有用です。これらの条件はどちらも、例えば物理学で関心のある多くの格子模型では満たされます。KQD は基底状態についてほとんど何もわかっていない場合に特に有用です。これは、KQD が本質的な収束保証を持ち、基底状態に関する知識が不十分なために代替手法が適用できないシナリオにも適用できるためです。
KQD は強力なツールですが、このプロトコルで時間を要する部分、特に射影ハミルトニアンの各要素の推定とクリロフ状態のオーバーラップの推定は改善の余地があります。別のアプローチとして、クリロフ法をサンプリングベースの手法と組み合わせる方法があり、これは次のレッスンのテーマです。
6. 付録
付録 I:実時間発展によるクリロフ部分空間
ユニタリ・クリロフ空間は次のように定義されます:
ここで は後で決定するタイムステップです。一時的に が偶数であると仮定し、 と定義します。上記のクリロフ空間にハミルトニアンを射影すると、それはクリロフ空間と区別がつかないことに注意してください:
つまり、すべての時間発展が タイムステップ分後ろにシフトされた空間です。 区別がつかない理由は、行列要素
が発展時間の全体的なシフトに対して不変であるためです。これは時間発展がハミルトニアンと可換であることから来ています。奇数の については、 の場合の解析を使えます。
このクリロフ空間のどこかに低エネルギー状態が必ず存在することを示したいと思います。[3] の定理 3.1 から導かれる以下の結果によってこれを示します:
命題 1: 関数 が存在して、ハミルトニアンのスペクトル範囲(基底状態エネルギーから最大エネルギーまで)内のエネルギー に対して...
- : から 離れた全ての に対して成立し、指数的に抑制されます。
- は に対する の線形結合です。
以下に証明を示しますが、完全で厳密な議論を理解したい場合以外は読み飛ばして構いません。ここでは上記の命題の含意に焦点を当てます。上記の性質 3 により、上記のシフトされたクリロフ空間が状態 を含むことがわかります。これが低エネルギー状態です。その理由を見るために、 をエネルギー固有基底で展開します:
ここで は 番目のエネルギー固有状態で、 は初期状態 におけるその振幅です。これを使うと、 は次のように表されます:
ここで、 が固有状態 に作用するとき を に置き換えられることを使いました。この状態のエネルギー誤差は
これを理解しやすい上界に変換するため、まず分子の和を の項と の項に分けます:
第一項を で上から抑えると:
ここで第一の不等号は和の各 に対して が成立することから、第二の不等号は分子の和が分母の和の部分集合であることから従います。第二項については、 であるため分母を で下から抑えます。すべてをまとめると、
残りの部分を簡略化するために、これらの に対して の定義から が成立することに注意します。さらに および で上から抑えると、
これは任意の に対して成立します。したがって を目標誤差に設定すれば、上記の誤差上界はクリロフ次元 に対して指数的に収束します。また の場合、上記の の項は完全に消えることにも注意してください。
議論を完結させるため、まず上記はクリロフ空間の最低エネルギー状態ではなく、特定の状態 のエネルギー誤差に過ぎないことに注意します。しかし、(レイリー・リッツ)変分原理により、クリロフ空間の最低エネルギー状態のエネルギー誤差は、クリロフ空間内の任意の状態のエネルギー誤差で上から抑えられます。したがって上記は、クリロフ量子対角化アルゴリズムの出力である最低エネルギー状態のエネルギー誤差の上界でもあります。
上記と同様の解析は、ノイズおよびノートブックで述べたしきい値処理手続きを追加的に考慮した形でも行えます。この解析については [2] および [4] を参照してください。
付録 II:命題 1 の証明
以下は主に [3] の定理 3.1 から導かれます。 とし、 を次数最大 の残差多項式(0 での値が 1 となる多項式)の空間とします。
の解は
であり、対応する最小値は
これを複素指数関数で自然に表せる形式に変換したいと思います。なぜなら、それが量子クリロフ空間を生成する実時間発展だからです。 そのために、ハミルトニアンのスペクトル範囲内のエネルギーを の範囲の数に変換する以下の変換を導入すると便利です:
ここで は を満たすタイムステップです。 であり、 が から離れるにつれて が増加することに注意してください。
次に、パラメータ 、、 を設定した多項式 を用いて、関数を定義します:
ここで は基底状態エネルギーです。 を代入すると、 が次数 の三角多項式、すなわち に対する の線形結合であることがわかります。さらに、上記の の定義から が成立し、 を満たすスペクトル範囲内の任意の に対して
参考文献
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).