HI-VQE Chemistry - Qunova ComputingによるQiskit Function
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
APIリファレンスを参照してください
Qiskit Functionsは、IBM Quantum® Premium Plan、Flex Plan、およびOn-Prem(IBM Quantum Platform API経由)Planのユーザーのみが利用できる実験的な機能です。プレビューリリースの状態であり、変更される可能性があります。
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
概要
量子化学において、電子構造問題は電子のシュレーディンガー方程式の解、つまり系の電子の振る舞いを記述する量子波動関数を見つけることに焦点を当てています。これらの波動関数は複素振幅のベクトルであり、各振幅は可能な電子配置の寄与に対応しています。
基底状態は系の最低エネルギー波動関数であり、分子系の研究において特別な重要性を持っています。基底状態を計算する最も正確なアプローチはすべての可能な電子配置を考慮しますが、配置の数が系のサイズとともに指数関数的に増加するため、より大きな系では扱いが困難になります。
Handover Iterative Variational Quantum Eigensolver(HI-VQE)は、分子系の基底状態を正確に推定するための革新的なハイブリッド量子古典手法です。量子ハードウェアと古典コンピューティングを統合し、量子プロセッサを使用して候補となる電子配置を効率的に探索し、古典コンピュータ上で得られた波動関数を計算します。コンパクトでありながら化学的に正確な波動関数を生成することで、HI-VQEは量子化学および材料科学における研究と発見を促進します。

HI-VQEは高精度で基底状態を効率的に推定することで、電子構造問題の計算複雑性を削減します。最も関連性の高い電子配置の中から慎重に選択されたサブセットに焦点を当て、精度と効率の両方を最適化します。
古典コンピュータと量子コンピュータの長所を組み合わせることで、HI-VQEは現在の推定波動関数を反復的に改良・改善します。その独自のサブスペース構築技術により、配置選択をより効率的にし、量子化学シミュレーションにおける計算制御と精度の向上をユーザーにもたらします。
アルゴリズムをより詳しく学びたい場合は、関連する研究論文を読んでください。
説明
分子系における電子配置の数は、系のサイズとともに指数関数的に増加します。ただし、基底状態などの特定の電子状態では、ごく一部の配置のみが状態のエネルギーに大きく寄与することが一般的です。選択配置相互作用(SCI)法はこのスパース性を利用して、最も関連性の高い配置を特定・集中することで計算コストを削減します。この配置のサブセットをサブスペースと呼びます。
HI-VQEは、分子系を表現するための量子コンピュータの固有の効率性を活用してサブスペース探索を支援します。電子構造問題を高精度で解くために、古典的および量子的サブルーチンを統合しています。既存の量子SCI手法とは異なり、HI-VQEは変分トレーニング、反復的なサブスペース構築、および対角化前の配置スクリーニングを組み合わせることで、量子測定、反復回数、および古典的対角化コストを削減することにより効率を向上させます。したがって、HI-VQEはより多くの量子ビットを必要とする大規模な分子系に適用でき、同程度の精度で問題を解くコストを削減します。
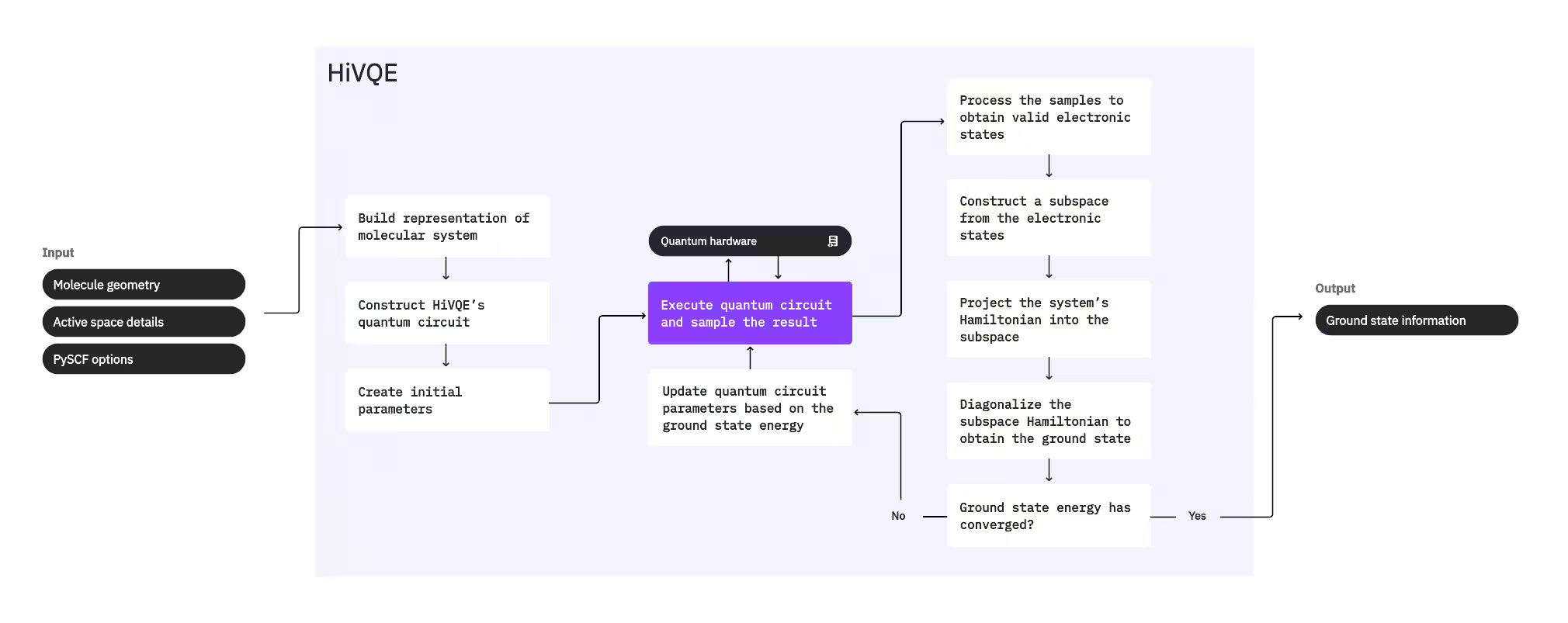
系の基底状態を計算するために、HI-VQEはまず古典的な化学パッケージPySCFを使用して、分子ジオメトリやその他の分子情報など、ユーザーが提供した入力から分子表現を生成します。次に、ハイブリッド量子古典最適化ループに入り、含まれる配置の数を最小化しながら基底状態を最適に表現するサブスペースを反復的に改良します。ループは、サブスペースサイズやエネルギーの安定性などの収束基準が満たされるまで継続し、その後、計算された基底状態波動関数とエネルギーが出力されます。これらの結果は、正確なポテンシャルエネルギー面の構築や系のさらなる分析に使用できます。
最適化ループは、量子回路のパラメータを調整して高品質なサブスペースを生成することに焦点を当てています。HI-VQEは3つの量子回路オプションを提供しています:excitation_preserving、efficient_su2、およびLUCJです。最適化はその一般的な適合性から、Hartree-Fock参照状態に近い状態で初期化されます。次に回路が量子デバイス上で実行され、得られた量子状態から配置がサンプリングされてバイナリ文字列として返されます。量子デバイスのノイズにより、サンプリングされた配置の一部は電子数やスピンを保存せず、物理的に無効な場合があります。HI-VQEはこれをqiskit-addon-sqdパッケージの配置回復プロセスを使用して対処しており、ユーザーは無効な配置を修正するか破棄するかを選択できます。
有効な配置は、寄与が最小限と予測されるものを除去するオプションのスクリーニングステップを経ます。これによりサブスペースの次元が削減され、対角化ステップのコストが低下します。スクリーニングが有効な場合、有効な配置から予備のサブスペースハミルトニアンが構築され、非常に緩やかな終了基準で対角化が実行されます。各配置の得られた振幅の精度は低いですが、この反復においてサブスペースから除外する配置を予測するのに効果的であり、高速に計算できます。
選択された配置はサブスペースに追加され、系のハミルトニアンがこのサブスペースに射影されます。サブスペースは反復的に更新され、最も関連性の高い配置が反復をまたいで保持されます。このアプローチは代替手法とは異なり、量子回路が各ステップで完全な基底状態を近似する必要がありません。
次に、サブスペースハミルトニアンが古典的に対角化され、最低固有値とその対応する固有ベクトルが取得されます。これらは基底状態とそのエネルギーの近似を表します。反復を通じてサブスペースの品質が向上するにつれて、計算された基底状態は真の基底状態をより良く近似します。この時点で、計算された基底状態に実質的な寄与をしない配置をサブスペースから除去するための追加のスクリーニングステップを実行できます。このステップにより、次の反復に引き継がれるサブスペースができる限りコンパクトになります。これは対角化から返される振幅に基づいて評価されます。これらの振幅は各配置の計算された基底状態への重要度の寄与を表しています。
収束チェックにより、さらなるトレーニングが結果を改善するかどうかが決定されます。改善する場合は、オプションの古典的拡張ステップが実行され、量子回路パラメータが更新されて計算されたエネルギーをさらに最小化し、プロセスが繰り返されます。古典的拡張ステップはサブスペースの追加配置を生成し、量子デバイスからサンプリングされた配置を補完します。まず対角化結果で最大の振幅を持つ配置を特定し、その後、特定された配置から一重および二重励起の新しい配置を生成します。これらの配置の所望の数がサブスペースに追加されます。
反復が収束したと判断されると、HI-VQEは計算された基底状態(サブスペース内の状態とその基底状態波動関数における振幅の形で)、そのエネルギー、および計算された状態が系のハミルトニアンの固有状態を形成しているかどうかの指標となるエネルギー分散の測定値を返します。
ユーザーは使用する量子回路と各量子回路のショット数を決定でき、サブスペースサイズを制御したり、量子生成された配置を補助するための配置の古典的生成を有効にしたりすることができます。このようにして、ユーザーはHI-VQEの動作を希望するアプリケーションに合わせて調整できます。
ライセンス
このQiskit Functionの使用は、ライセンスを取得して上限を引き上げない限り、最大20量子ビットを必要とする問題に限定されています。
ライセンスの取得についてのお問い合わせは、qiskit.support@qunovacomputing.comまでメールをお送りください。
はじめに
まず、functionへのアクセスをリクエストしてください。 次に、IBM Quantum® APIキーを使用して認証し、すでにローカル環境にアカウントを保存していると仮定して、Qiskit Functionを次のように選択してください:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
例
最初の例は、HI-VQEアルゴリズムを使用してNH3分子の基底状態エネルギーを計算する方法を示しています。
分子ジオメトリとオプションの定義
NH3の分子ジオメトリは、各原子を「;」で区切った直交座標で提供されます。
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
追加のオプションを分子系のために以下の辞書形式で定義して提供できます。
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
ジオメトリとオプション入力を使用してfunctionを実行します。
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
問題が発生した場合のサポートリクエストで提供できるように、Function job IDを印刷しておくことをお勧めします。
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
この例では、NH3分子のsto3g基底で8軌道を持つ16量子ビットを使用します。 Qiskit Functionワークロードのステータスを確認したり、次のように結果を返したりすることができます:
print(job.status())
QUEUED
ジョブが完了したら、result()インスタンスで結果を取得できます。
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
基底状態エネルギーにアクセスするには、「energy」キーを使用します。「eigenvector」キーは、結果の「states」に格納された電子配置のビット文字列表記に対応するCI係数を提供します。
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
パフォーマンス
このセクションでは、Li2Sの24量子ビットケース、N2分子の40量子ビットケース、およびFeP-NOシステムの44量子ビットケースに対するHI-VQEのベンチマーク計算の実証を示します。
24量子ビットのLi2S分子の解離ポテンシャルエネルギー面曲線
PES曲線はFCI参照値とRHFからの初期推定値とともに示され、FCI参照値からのエネルギー誤差も示されています。
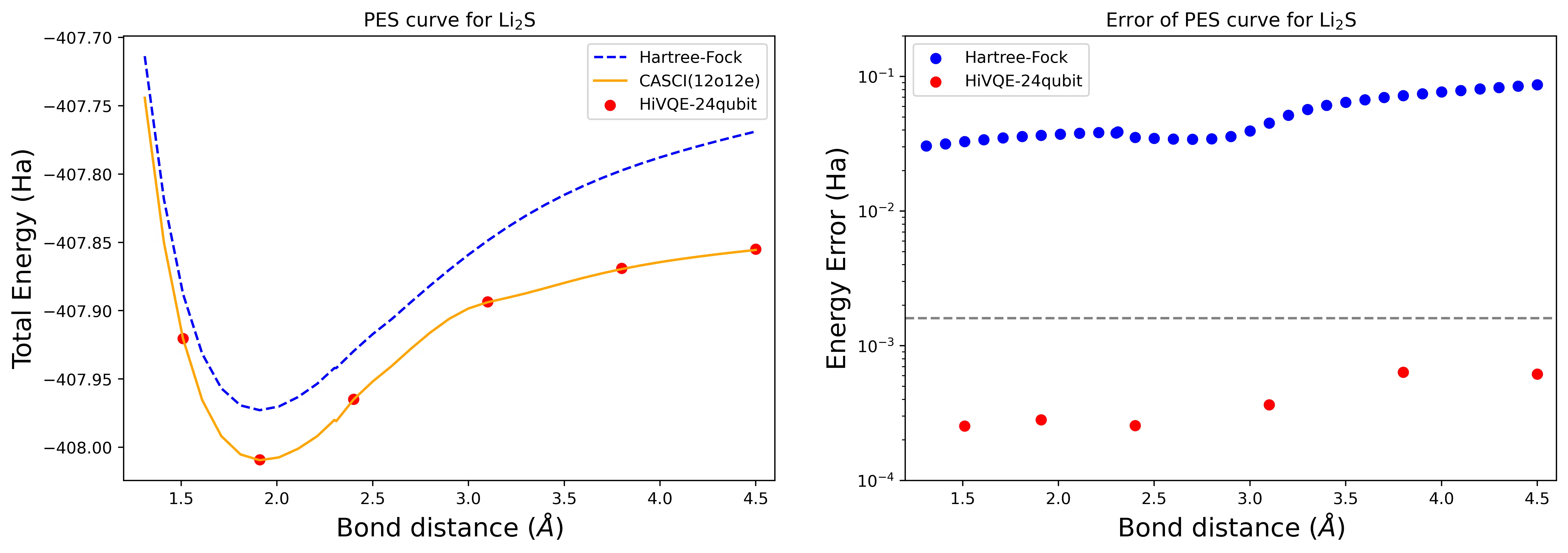
計算は以下のジオメトリとオプションで実施されています。
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
赤い点はHI-VQE計算結果(6つの異なるジオメトリ)を表しており、1.51、2.40、3.80オングストロームに対応する3つのジオメトリが上のセルで入力として提供されています。
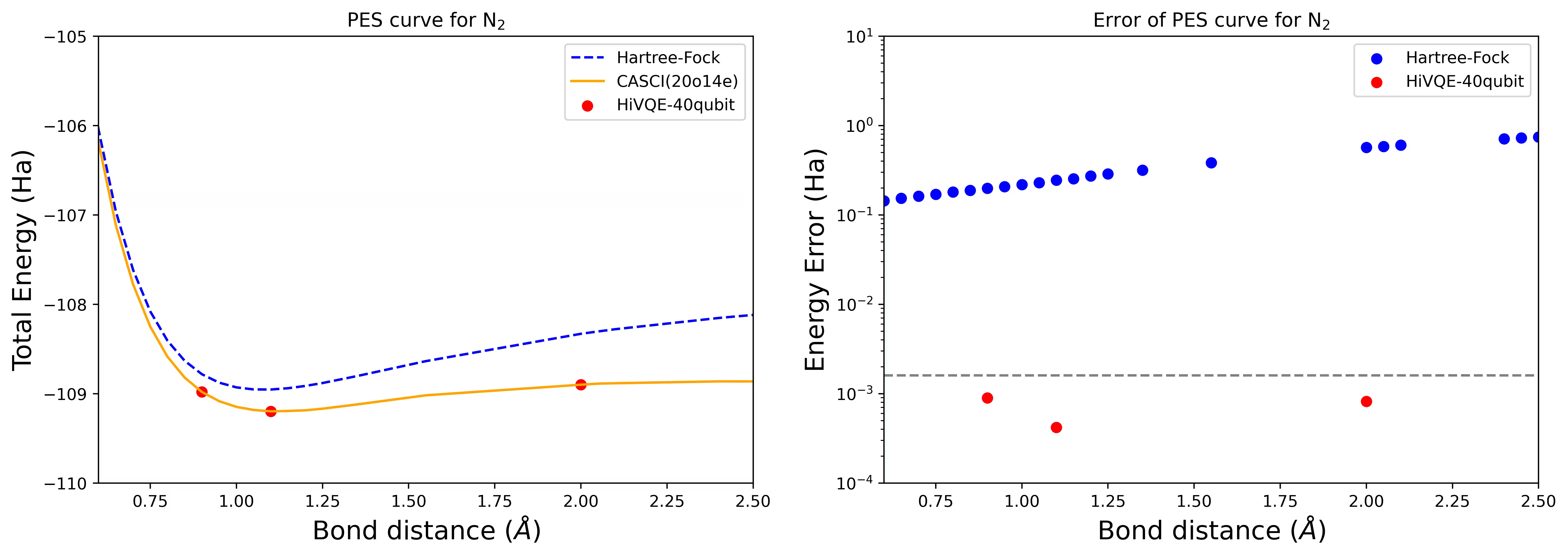
40量子ビットのN2分子の解離PES曲線
窒素分子は、Hartree-Fock状態を超えた大きな相関エネルギー寄与を持つ多参照系として特定されています。cc-pvdz基底、homo-lumo活性軌道選択を使用した(20o,14e)のN2分子のベンチマーク計算を実施しました。この問題を表現するための完全活性空間(CAS)の数は6,009,350,400です。強力なデスクトップコンピュータ(16cpu/64GB)ではこの数の状態での固有値問題(エネルギーと電子構造)の解を得ることは不可能です。HI-VQEを使用することで、ユーザーはCAS状態のサブスペースを効率的に探索して化学的に正確な結果を見つけながら、計算リソースを大幅に節約できます。以下のプロットはN2分子解離の40量子ビットHI-VQE計算のPES曲線を示しています。
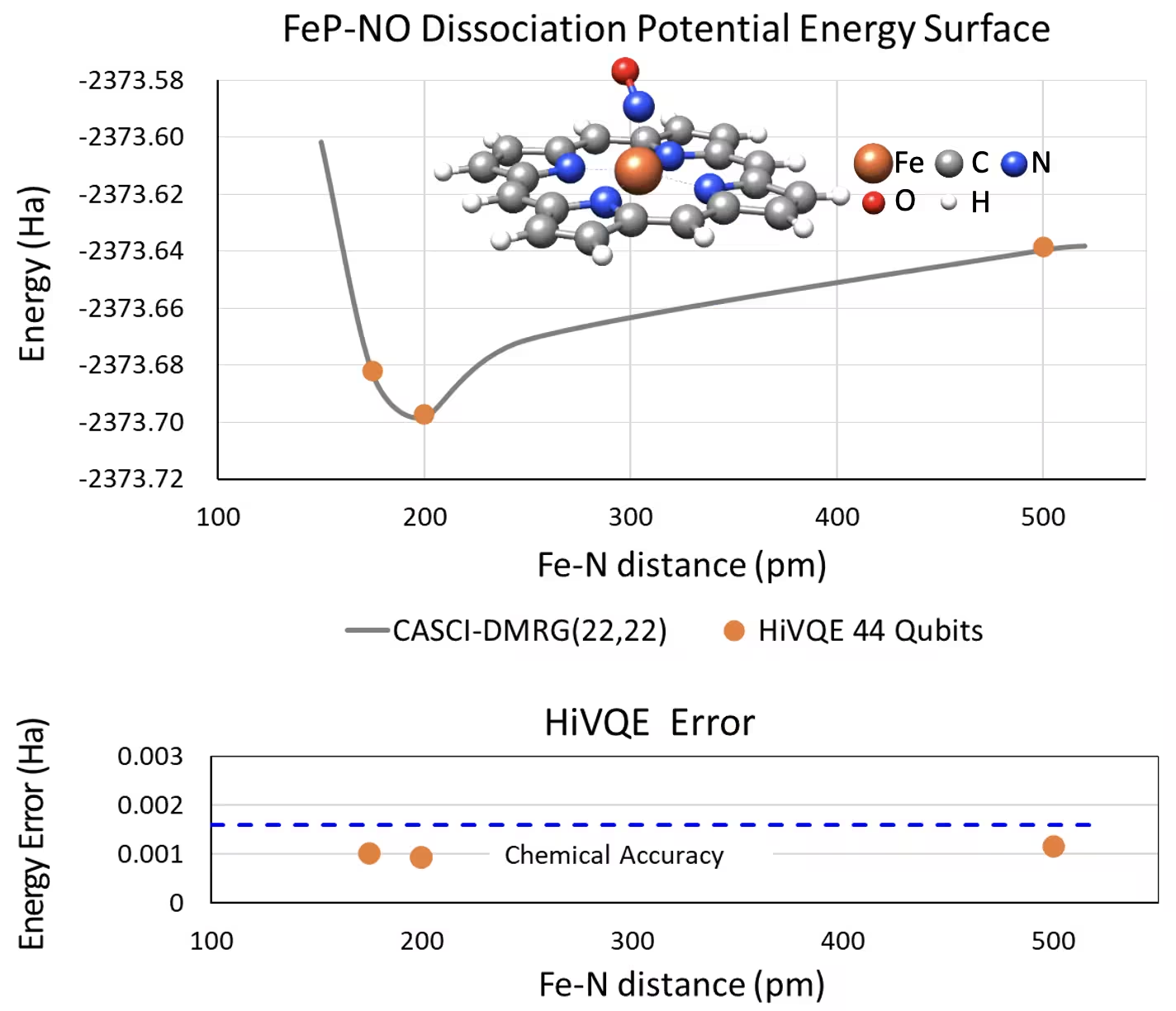
44量子ビットのNOシステムを持つ五配位鉄(II)-ポルフィリンの解離PES曲線
もう一つの興味深い化学系は、配位した一酸化窒素(NO)リガンドを持つ鉄(II)-ポルフィリン(FeP)錯体です。これは様々な生理的プロセスで重要な役割を果たす生物学的に関連したメタロポルフィリン系を表しています。この例では、HI-VQEを使用してFePとNO間の分子間相互作用の正確なポテンシャルエネルギー面曲線(異なる分離ジオメトリに対する基底状態エネルギー)を推定しています。組み合わせた系は合計6-31g(d)基底で450軌道と202電子(450o,202e)を持ちます。Homo-lumo活性軌道選択を使用して実際のケースから小さいケース(22o,22e)を計算しました。以下のベンチマーク結果から、CASCI(DMRG)(22o,22e)参照という最先端の古典コンピュータ化学計算と比較して化学精度(> 1.6 mHa)を達成できました。
ベンチマーク
- 正確な行列サイズは、FCIやCASCIなどの正確な解に対する行列式の数です。
- HI-VQE計算はそのサブスペースをサンプリングして計算します(HI-VQE行列サイズ)。
- 合計時間にはQPUランタイムとQiskit FunctionのCPU実行時間が含まれます。
- 精度は正確な解からのエネルギー差から推定されます。
| 化学系 | 量子ビット数 | 正確な行列サイズ | HI-VQE行列サイズ | 正確な解からのE(diff)(mHa) | 反復回数 | 合計時間 | QPUランタイム使用量 |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
エラーメッセージの取得
ワークロードが失敗した場合、ステータスはERRORとなり、job.result()を呼び出すと例外が発生します:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
サポートを受ける
このfunctionについてサポートが必要な場合は、qiskit.support@qunovacomputing.comにメールを送ってください。
特定のエラーのトラブルシューティングについてサポートが必要な場合は、エラーが発生したジョブのFunction job IDを提供してください。
次のステップ
- このフォームに記入してfunctionへのアクセスをリクエストしてください。
- このQiskit FunctionのAPIリファレンスを参照してください。
- HI-VQEを使用したFeP-NOの解離PES曲線の計算チュートリアルを試してください。
- Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.を読んでください。
- Qunova HiVQEによる解離PES曲線チュートリアルを試してください。