マッピング
Olivia Lanes によるマッピングの動画をご覧いただくか、YouTube で別ウィンドウとして開いてください。
はじめに
このレッスンでは、量子プログラムを定義する際の第一歩であり、最も困難なことが多いステップ、すなわち量子コンピュータ上で実行する問題のマッピングに焦点を当てます。このステップでは、ユーザーが計算上の問題からスタートし、量子コンピュータで解けるものへと変換する方法を扱います。
このコースのレッスン 2 と 3 では、マッピング段階が Qiskit パターンフレームワークにおける全 4 ステップの最初のステップであることに触れました。それらのレッスンから、マッピングの目標は計算上の問題を、量子コンピュータを使って評価できるコスト関数または期待値へと変換・書き換えることだと覚えているかもしれません。
レッスン 3 では、組み合わせ最適化において計算が難しいながらも非常に一般的な問題である Max-Cut を具体例として取り上げました。そこでは、最初のグラフ問題を量子コンピュータで解けるものへと変換するためのいくつかのステップを順に進めました。グラフにおける最大カット数を求める問題をコスト関数へと変換し、そのコスト関数をハミルトニアンとして書き直し、基底状態が最大カットに対応する試行量子状態を準備しました。最後に、目的の試行量子状態を表す量子回路を構築し、状態が時間発展できるように特定のゲートを追加しました。 この一連のステップはすべてマッピングの一部です。具体的なステップは Max-Cut 問題に固有のものでしたが、同じ一般的な手順は量子化学や量子シミュレーションなど、他の多くのアプリケーションにも適用できます。
マッピングは難しい場合があります。すべての問題に対応する一律の戦略は存在しないため、とっつきにくく感じることもあります。このレッスンでは、マッピングにおける一般的な考慮事項を確認した上で、代表的な問題例を通じて、量子コンピュータへの問題のマッピング方法をさまざまな角度からご紹介します。
一般的な考慮事項
問題を量子コンピュータにマッピングするために使う具体的な戦略は問題によって異なりますが、おおむね 2 つの主要な目的を達成します。
まず、問題を実現可能にするために単純化する必要があるかもしれません。これは量子に限ったことではありません。あらゆる科学分野が、無関係な細部を無視しながら関心のある現象を研究するために、単純化されたモデルを使用しています。物理学では、この原則を極端に推し進めた有名な表現があります:「球状の牛を仮定せよ」。系をそのままの姿で正確に記述することは難しすぎますが、代わりに合理的な単純化を行うことで、依然として有益な解が得られます。量子コンピューティングにおいてこれを行う方法の例として、ハードウェア効率の良いアンザッツを選択してサイズや回路の深さを制限すること、複雑な時間発展を打ち切ること、量子状態の最終エネルギーへの寄与が少ないハミルトニアン項を無視すること、などが挙げられます。
次に、マッピングでは量子コンピュータが理解できる形で問題を記述することが必要です。多くの場合、以下の 3 つの問いかけが伴います:
- モデルにおいて、量子ビットは何を表しますか?
- 問題は連続的ですか?量子コンピュータはデジタルですので、問題が連続的である場合は離散化する方法を見つける必要があります。
- 最後に、問題のトポロジーはハードウェアのトポロジーと一致していますか?一致していない場合は、うまく機能させるためにいくつかの工夫を実装する必要があるかもしれません。
最初の問いに取り組みましょう。これはマッピングを理解する上での難しさの核心にある問いです:量子ビットは何を表すことができるのでしょうか?
量子ビットはさまざまなものを表すために使用できます。最初の、そしておそらく最も単純な例は、グラフ上のノードです。グラフはさまざまな数学的問題における接続性を示すために使われており、ノードはネットワーク内の点や実体を表す基本的な要素です。ネットワーク全体が何を表すかによって、それは都市、人、または格子の中の強磁性体になり得ます。
量子ビットはボゾンやフェルミオンを表すためにも使用できますが、1 つの量子ビットが 1 つのボゾンや 1 つのフェルミオンに正確に等しいわけではないことに注意が必要です。レッスンの中でさらに詳しく説明しますが、少し複雑です。
ここで、少し複雑な例を見ていきます。これらのモデルでは、単一の量子ビットで語ることはもはや意味をなさず、代わりに物理的な何かを構成するためにグループが必要になります。例えば、ヘビーヘキサゴナルトポロジーで表された量子ビットのグループを使用して、アミノ酸の幾何学的位置(ポリマー鎖)を表すことができます。別の例として、高エネルギー物理学モデルにおけるハドロン散乱のシミュレーションがあり、これはハドロン波束の時間発展をシミュレーションすることで実現できます。この場合、量子ビットのレジスタを使って量子場の異なる状態(その場の真空状態、または真空上を伝播する波束)をエンコードできます。
しかし、ここまでは抽象的な話にとどめてきました。これらの例を詳しく見ていきましょう。
マッピングの例
Max-Cut
最初の例から始めましょう。最も単純なマッピング問題の一つは、すでにある程度詳しく扱った Max-Cut の例です。この問題では、1 つの量子ビットがグラフ上の 1 つのノードに対応するため、マッピングは非常に簡単でした。
Max-Cut 問題をマッピングするために、QUBO 定式化を使ってコスト関数をハミルトニアンとして表現したことを思い出してください。ハミルトニアンコスト関数とは、ハミルトニアンの基底状態に問題の最適解をエンコードする関数です。コストハミルトニアンを構築するために、Qiskit の SparsePauliOp クラスを使ってグラフの接続性を指定し、量子演算子へのマッピング段階が完了しました。そして量子回路は単純に QAOA アンザッツでした。復習のために、実用規模の QAOA レッスンを参照してください。そこで詳細をより丁寧に解説しています。
そのレッスンでは、100 量子ビットの実用規模の例においても、グラフの接続性がすでに超伝導ハードウェアのトポロジーと一致していました。そのため、異なるトポロジーへの対処方法を考える必要はありませんでした。しかし、常にそうとは限りません。これまで取り上げた例よりも複雑または密に接続されたグラフの場合は、ハードウェアの実効的な接続性を変更するために一連の SWAP ゲートを実装する必要があります。これは Qiskit パターンの第 2 ステップであるトランスパイルで処理されますが、マッピングの段階でも念頭に置いておく必要があります。
タンパク質折り畳み
次に、IBM® およびニューサウスウェールズ大学の研究者たちが執筆した「Resource-efficient quantum algorithm for protein folding(タンパク質折り畳みのためのリソース効率の良い量子アルゴリズム)」と題した論文でモデル化された例を探ってみましょう。
生化学の背景を少し説明します:タンパク質は長いアミノ酸の鎖から構成される高分子です。これらの鎖は折り畳まれて複雑な構造を形成し、幅広い生物学的機能を果たします。タンパク質の三次元空間における構造を決定し、構造と機能の関係を理解することは、今日の生化学において最も難しい問題の一つです。タンパク質は、アミノ酸間の相互作用によって有用な構造に折り畳まれます。構造がねじれ、折り畳まれるにつれて、鎖に沿って互いに遠く離れたアミノ酸が隣り合う位置に来て、強く相互作用することがあります。
これを量子コンピュータでモデル化するには、アミノ酸間のすべての相互作用を記述するハミルトニアンが必要です。そして、ハミルトニアンのエネルギーを最小化する状態を見つけることで、最終的な構造を予測できます。ここでは、アミノ酸鎖を量子コンピュータでどのようにモデル化するか、そして相互作用エネルギー計算のためのアミノ酸間距離をどのように取得するかに焦点を当てます。これにより、量子コンピュータでシミュレーションするために必要なハミルトニアンへの必要な貢献をすべて収集できます。
実際のタンパク質では、アミノ酸は連続した可能性のある位置を占めることができます。しかし、格子モデルを使って単純化し、各アミノ酸がグリッド上の一点を占めるよう制限します。ここでは、著者たちは四面体格子を使用しました。クイックノート:ここでは Max-Cut 問題のようにノード自体ではなく、エッジの方向をエンコードしています。各量子ビットは四面体グリッドに沿った一ステップの経路を表します。格子内での方向が異なるため、隣接するサイトは A または B としてラベル付けされています。
タンパク質鎖はこの格子上の一連の曲がりや方向として表されます。アミノ酸間の各曲がりは 4 つの方向のうちの 1 つを取ることができ、それは四面体のエッジに対応します。これら 4 つの可能な曲がりは、4 つの量子ビットを使って状態 0001、0010、0100、または 1000 にエンコードされます。
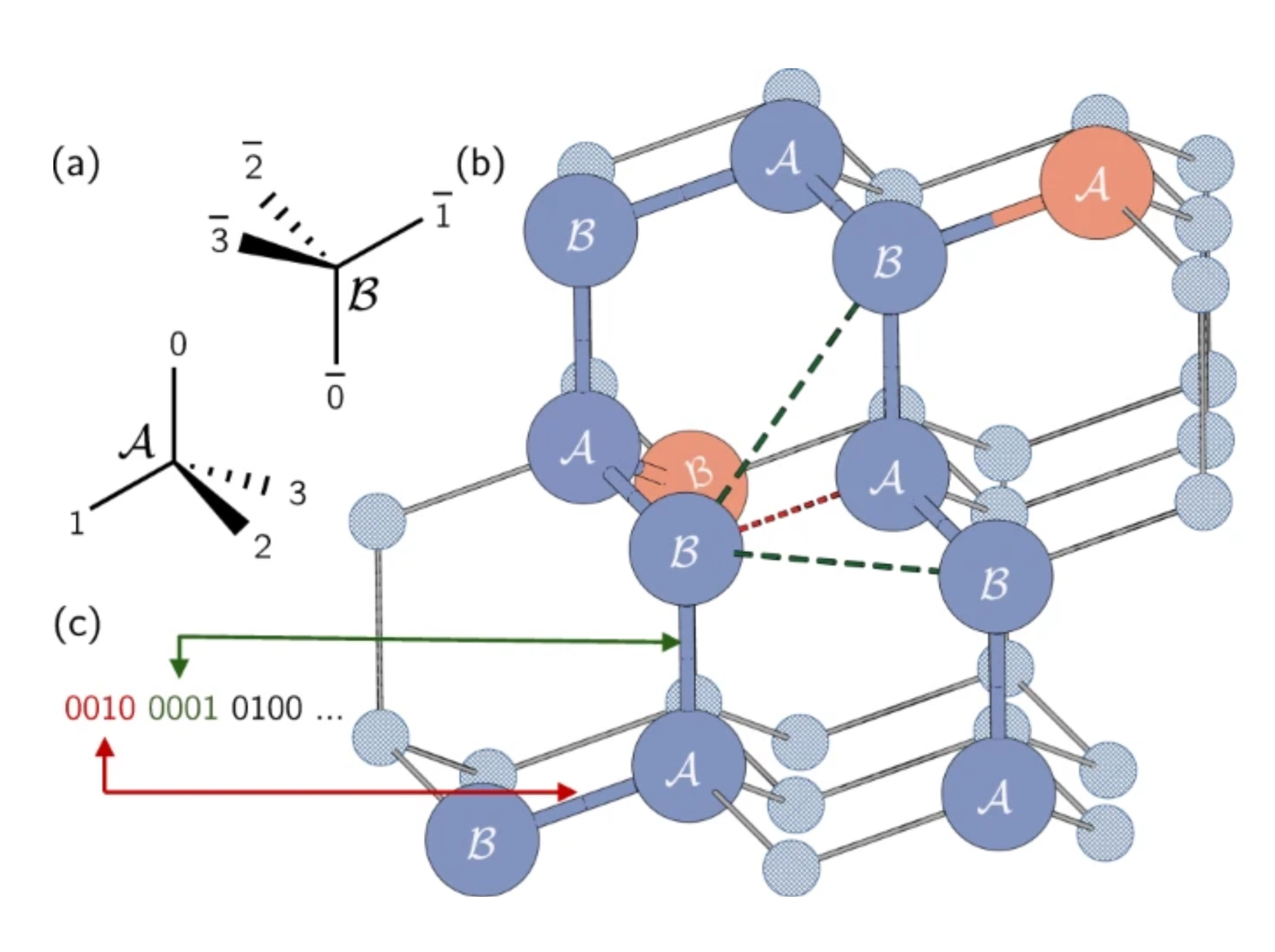
上の図の例を見てみましょう。四面体格子の赤丸で囲まれた「B」とラベルされた点に最初のアミノ酸を置きましょう。最初のアミノ酸から 2 番目への方向は任意です。なぜなら、系は常に回転させてそのエッジを任意の方向に向けることができるからです。したがって、最初の点の下の「A」とラベルされた点に 2 番目のアミノ酸を置くことができます。わかりにくいかもしれませんが、2 番目から 3 番目への経路も任意です。3 つの選択肢すべてが、2 本のエッジの間に約 109.5 度の角度を持つ結果になります。この 2 番目のエッジを選ぶことは、単に空間内でのタンパク質の向きを決めるだけです。したがって、一般性を失うことなく、最初の 2 つの曲がりを単に 0001 と 0010 とすることができます。
これらの単純化により、アミノ酸鎖の配置はこの式で与えられます:
ここまで、四面体のエッジを量子ビットにマッピングし、量子回路はアンザッツになります。次に、基底状態が最適な折り畳みパターンを与えるように、問題のエネルギーをハミルトニアンにエンコードする方法を考える必要があります。
任意の配置に対して、アミノ酸間の相互作用による関連エネルギーが存在します。これらの相互作用は、2 つのアミノ酸が互いに近いときに最も強くなります。明らかに、鎖の主鎖で隣接するアミノ酸は常に相互作用します。しかし、タンパク質はねじれたり折り畳まれたりできるため、他のアミノ酸ペアも相互作用することがあります。例えば、タンパク質が折り畳まれた後、アミノ酸 10 と 20 が隣接したサイトになることがあります。したがって、配置の量子ビットにエンコードされた情報を使って、アミノ酸 と の間の距離 を記述する式が必要です。それにより、分離距離を使ってどの程度強く相互作用しているかを決定できます。
まず、エッジ がアミノ酸 での曲がりに使われているかどうかを示す関数 を導入しましょう。ここで は 4 つの可能な方向のいずれかを取ることができます。上で始めた配置に基づいて、これらの関数を書くことができます:
そして、インデックス から までの A 格子と B 格子における ラベルの曲がりの数の差を として定義できます:
なぜこのようにするのでしょうか?実は、格子サイト A から B への の曲がりは、格子サイト B から A への の曲がりによって正確に打ち消されます。つまり、サイト のアミノ酸からサイト のアミノ酸までの距離を知るには、A サイトと B サイトから エッジに沿って行ったステップの差を求めるだけでよいのです。A サイトと B サイトはタンパク質主鎖に沿って必ず交互に現れるため、これは によって捉えられます。この同じ議論は 4 つのエッジ型すべてに適用されます。したがって、四面体格子ステップでのアミノ酸間の総距離はこの式で計算できます:
しかし、アミノ酸間の総距離のこの長い方程式からどのようにしてハミルトニアンを得るのでしょうか?まず、簡単な幾何学を使って格子ステップの距離からユークリッド空間の距離に変換できます:
次に、これらの距離はタンパク質配置のエネルギーを計算するために使用されます。目的に応じて、ペアが相互作用していると見なすカットオフ距離を設定するか、より複雑な方法を選択するかもしれません。
明らかではないかもしれませんが、これを行うことでマッピング段階は実際に完了しています。量子ビットの状態は各格子サイトでのタンパク質の「曲がり」を示し、曲がりの集合が任意のアミノ酸ペア間の距離を決定します。さまざまな種類のアミノ酸ペアは異なる相互作用を持ち、引力のものも斥力のものもあります。配置と距離を使って既知のアミノ酸相互作用が「オン」か「オフ」かを判断するだけであれば、これらの強度はすでに算出されており、次のような表から単純に参照できます:
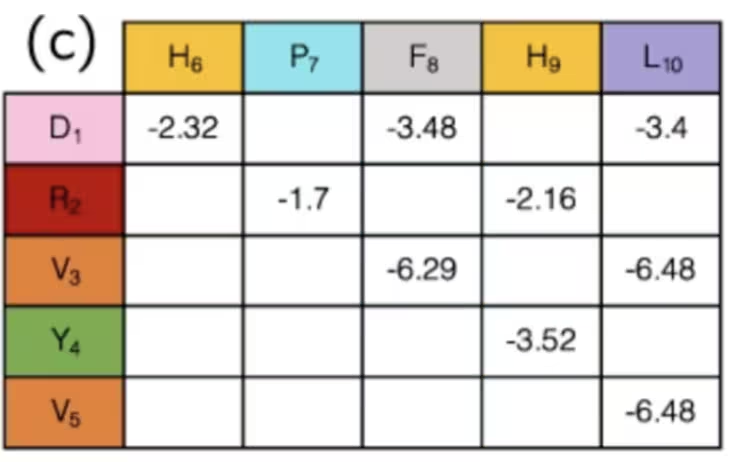
まとめると、この例では量子ビットは格子上の経路のステップを示すために使用され、それらが合わさってアミノ酸の鎖を形成します。折り畳みや曲がり方をシミュレーションすることで、医学研究でより良い成果が得られることが期待されます。 このハミルトニアンのいくつかの項の計算方法については省略しました。それらはこの問題に非常に特有のものでしたが、格子上の方向を定義することはより広く応用できます。一般的なハミルトニアンが得られたら、量子コンピュータにネイティブなパウリ演算子へと変換することが常に必要になります。それが次に議論することです。
ジョルダン・ウィグナー変換
次に、素粒子の系をパウリ演算子に変換する方法を探ってみましょう。
素粒子はボゾンとフェルミオンの 2 つのカテゴリに分けられます。光子やヒッグス粒子のようなボゾンは、ある統計的ルールに従います。電子やニュートリノのようなフェルミオンは、別の統計的ルールに従います。両者の主な違いは、ボゾンは同じ状態を占めることができる点です。基底状態や任意の励起状態に何個でもボゾンが存在できます。一方、フェルミオンは排他的で、すべての粒子が独自の量子状態を持つことを要求します。
ボゾンは整数スピンを持ち、フェルミオンはスピン 1/2 の電子やより珍しいスピン 3/2 の粒子のような半整数スピンを持ちます。粒子の系を記述するには、エネルギーの記述が必要です。フェルミオンに焦点を当てましょう。c 演算子 と呼ばれるもので書かれたハミルトニアンから始めることができます。これらは基本的に、系の状態からフェルミオンを生成または消滅させることに対応する数学的オブジェクトです。これらはしばしば および として書かれ、 は状態 にフェルミオンを生成する演算子、 は状態 のフェルミオンを消滅させる演算子です。
しかし、量子コンピュータは通常、フェルミオン系を表現するための特定のルールを持つ量子ビット基底で動作します。フェルミオン演算子の言語では固有に動作しません。このギャップを埋めるために、このフェルミオン表記を量子ゲートに自然に対応するパウリ演算子にマッピングする必要があります。
フェルミオンのためのいくつかのそのような変換があります。一般的な選択肢はジョルダン・ウィグナー変換です。ブラビ・キタエフ変換とパリティマッピングはより最近のフェルミオンエンコーディングです。ボゾン演算子は、ホルスタイン・プリマコフ変換を使用するか、フォック状態を利用可能なボゾンモードのサブ基底に直接マッピングすること、または他のオプションによって変換できます。他のエンコーディングも活発に研究されています。今のところ、ジョルダン・ウィグナー変換だけに焦点を当てます。
ジョルダン・ウィグナー変換は、1 つのフェルミオンを多数の量子ビットにマッピングすることを含みます。なぜ各電子を表すために 1 つの量子ビットを割り当てないのでしょうか?これは同一フェルミオンの識別不可能性に関係しています。量子ビットは識別可能で、電子は識別できません。例えば、任意のデバイス上の個別の量子ビットを簡単にラベル付けして識別することができます。しかし、電子の識別不可能性は、電子にラベルをつけることができないことを意味します。したがって、特定の電子ではなく、1s、2p、2p などの占有された軌道に従って演算子をラベル付けする必要があります。つまり、各量子ビットは分子内の軌道の役割をほぼ果たし、それが占有されているか非占有であるかを表します。ただし、その方法は少し複雑です。ジョルダン・ウィグナーマッピングは反対称性を追跡し、フェルミオン系全体の正しい統計を確保します。 ジョルダン・ウィグナーマッピングは、これらの関係を使用してフェルミオン演算子をパウリ演算子で表現します:
ジョルダン・ウィグナーマッピングは、軌道と量子ビットの一対一対応があるため、概念的に単純です。パリティマッピングを含む同様の目標を達成する他のマッピングもあります。状態のパリティの計算には複数の量子ビットの考慮が必要です。パリティマッピング(および一部の他のマッピング)では、1 つの量子ビットが 1 つの軌道に対応するという解釈は成立しません。 では、簡単な例を見ていきましょう。単一量子ビットの相互作用 を計算したいとします。生成・消滅演算子の定義を代入することから始めます。
交換子 です。したがって、最終的な式は次のようになります:
これにより、フェルミオン式を量子コンピュータが理解できるパウリ演算子で書き直すことに成功しました。
Qiskit でジョルダン・ウィグナーマッピングを実装する方法を簡単に説明しましょう。このような変換がどのように機能するかを理解することは重要ですが、特に大規模なシステムでは毎回手作業で計算するのは現実的ではありません。幸いにも、Qiskit は SparsePauliOp 関数を使ってこれを簡単にしてくれます。
おおまかに言えば、手順は次のとおりです:
- SparsePauliOp の
from_list関数を使って、マッピングするパラメータ空間のサイズに対応する恒等演算子を作成します。 - 先に示した生成・消滅演算子の定義に従い、SparsePauliOp の
from_list関数を使って 、、 パウリ演算子を定義します。これにより、フェルミオンの生成・消滅演算子が量子ビットのスピン演算子にマッピングされ、フェルミオン占有数が量子ビットの計算基底にエンコードされます。 - 上記の演算子を目的の軌道に適用することで、パウリ基底において目的のハミルトニアンを生成します。これは通常、コア(非相互作用)軌道を表す恒等行列の作成に対応し、その後、問題の具体的な内容に対応する重みを持つ活性空間に生成・消滅演算子を適用します。
ジョルダン・ウィグナーマッピングの仕組みを完全に理解したところで、より複雑な例を見てみましょう。前のレッスンで取り上げた「Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits(デジタル量子コンピュータ上での基底状態準備のためのスケーラブル回路:100 量子ビットにおけるシュウィンガーモデルの真空)」という論文を覚えているかもしれません。論文の詳細には改めて立ち入りません。電場がない場合に サイトを持つ格子に対するシュウィンガーモデルのハミルトニアンを表現するために使用されているジョルダン・ウィグナーマッピングだけに焦点を当てます。
ここでは、1 つの量子ビットがこのモデルで何を表すかを具体的に特定することがかなり難しいです。なぜなら、量子ビットの集合全体が物理的なもの(この場合は波束)を構成するからです。代わりに、量子ビットをおよそ空間の離散化された断片として考えることができます。ここで、 は格子の体積であり、各要素(単位格子)は 2 つの量子ビットを含みます。前のスライドで見たフェルミオン演算子は、特定のサイトにおける波動関数の振幅を記述します。したがって、ハミルトニアンにはこれらのフェルミオン生成・消滅演算子が含まれます。そのため、ジョルダン・ウィグナー変換を使ってこれらの演算子をパウリ演算子にマッピングします。
ここで はパウリ演算子 であり、 は です。このフォーマットで書かれたハミルトニアンが得られると、マッピング段階の難しい部分は終わり、パウリ演算子を使って回路に簡単に書き込めるようになります。
まとめ
最も単純なものから始まり、ジョルダン・ウィグナー変換をハドロン動力学に適用するところまで、近年の量子コンピューティング分野で使用されてきた具体的なマッピング手法の 4 つの例について説明しました。 この資料の多くは非常に技術的なものであり、初めて触れる場合は非常に難しく感じることがあるかもしれません。しかし、練習に時間をかけるほど慣れていきます。これがこのコースが「実践の中の量子コンピューティング」と呼ばれる理由です。最初から誰もがすぐに習得できるものではありません。学習し、考え込み、おそらくは苦しむ時間も必要です。しかし、その不快感の中にじっくりと留まり、学びを続ける中で生じる疑問を真剣に探求することをお勧めします。それが唯一の学習方法なのです。