変分量子固有値ソルバー(VQE)
このモジュールを受講するには、Pythonの動作環境と、以下の最新バージョンのパッケージがインストールされている必要があります:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
これらのパッケージのセットアップとインストール方法については、Qiskitのインストールガイドを参照してください。実際の量子コンピューターでジョブを実行するには、IBMクラウドアカウントの設定ガイドの手順に従ってIBM Cloudアカウントを作成する必要があります。
このモジュールはテスト済みで、QPU時間を約8分使用します。これは目安であり、実際の使用量は異なる場合があります。
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
はじめに
20世紀初頭に量子力学モデルが発展して以来、科学者たちは電子が原子核の周りを一定の軌道に沿って動くのではなく、軌道(オービタル)と呼ばれる確率の領域に存在することを理解してきました。これらの軌道は、電子が占有できる特定の離散的なエネルギー準位に対応しています。電子は自然に最も低いエネルギー準位(基底状態)に存在しますが、十分なエネルギーを吸収すると、より高いエネルギー準位(励起状態)へ遷移することができます。励起状態は一時的なものであり、電子はやがて低いエネルギー準位へと戻り、吸収したエネルギーを(多くの場合、光の形で)放出します。このエネルギーの吸収と放出という基本的なプロセスは、原子がどのように相互作用し、結合を形成するかを理解するうえで重要です。
原子が集まって分子を形成する際、原子軌道が結合して分子軌道が生まれます。これらの分子軌道における電子の配置とエネルギー準位が、生成される分子の特性と化学結合の強さを決定します。たとえば、2つの水素原子から水素分子()が形成される場合、各原子の電子は原子軌道を占有しています。原子が互いに近づくと、これらの原子軌道が重なり合い、新たな分子軌道が形成されます。一方はエネルギーが低い結合性軌道、もう一方はエネルギーが高い反結合性軌道です。各水素原子から1つずつ計2つの電子は、優先的に低エネルギーの結合性軌道を占め、分子を安定した共有結合で結びつけます。分離した原子と形成された分子のエネルギー差、特に分子軌道における電子のエネルギーが、結合の安定性と特性を決定します。
以下のセクションでは、分子に焦点を当て、この分子形成プロセスを探求します。実際の量子コンピューターと古典的な最適化手法を組み合わせて、この単純ながら基本的なプロセスのエネルギーを求めます。この実験は、量子計算が計算化学の問題解決にどのように応用され、電子エネルギーの役割についての洞察を提供できるかを実践的に示すものです。
VQE — 固有値問題のための変分量子アルゴリズム
化学における近似手法 — 変分原理と基底関数系
エルヴィン・シュレーディンガーの量子力学への貢献は、新しい電子モデルの導入にとどまりません。根本的には、彼は有名な時間依存シュレーディンガー方程式を導出することで波動力学を確立しました:
ここで、はハミルトニアン演算子であり、系の全エネルギーを表します。また、は系の量子状態に関するすべての情報を含む波動関数です。(注:は全時間微分であり、ここではエネルギー固有値を陽に含めていません。)
ただし、原子や分子の許容されるエネルギー準位を決定するような多くの実用的な応用では、時間依存の形から定常状態を仮定することで導出される時間非依存シュレーディンガー方程式(エネルギー固有値方程式)を代わりに使用します。定常状態とは、空間上の特定の点で粒子が見つかる確率密度が時間とともに変化しない量子状態のことです。
この形式では、は量子状態に対応するエネルギー固有値を表します。ハミルトニアンには、電子と原子核の運動エネルギー、電子と原子核の間の引力、電子間の斥力など、さまざまなエネルギーの寄与が含まれています。
エネルギー固有値方程式を解くことで、原子・分子系の量子化されたエネルギー準位を計算できます。しかし、分子の場合、電子の空間的な分布を記述する波動関数が複雑かつ高次元であるため、厳密に解くことは困難です。
そのため、科学者たちは実用的かつ正確な解を得るために近似手法を用います。本研究では、2つの主要な方法に焦点を当てます:
-
変分原理
この方法は波動関数を近似し、系の目標エネルギー(通常は基底状態エネルギー)にできるだけ近づくように調整します。変分原理の背後にある重要な考え方は単純です:
- 波動関数(「試行関数」)を推定すると、そこから計算されるエネルギーは常に系の基底状態エネルギー()以上になります。
- 試行関数のパラメーター、すなわちを調整することで、基底状態エネルギーのより良い近似を得ることができます。
- その精度は、試行波動関数の選択に大きく依存します。適切でない試行関数を選ぶと、エネルギーの推定値が正確な値から大きく外れる可能性があります。
-
基底関数系の近似
第2の近似手法は、波動関数を構築する段階における基底関数系のアプローチです。量子化学では、分子に対してシュレーディンガー方程式を厳密に解くことはほぼ不可能です。代わりに、複雑な多電子波動関数を、あらかじめ定義されたよりシンプルな数学的関数を組み合わせることで近似します。基底関数系とは本質的に、分子中の原子を中心に配置されたこれらの既知の数学的関数の集合であり、系における電子の形状と振る舞いを表現するための構成要素として使用されます。詳細な彫刻を標準的なレゴブロックだけで再現しようとするようなものと考えてください。ブロックの種類やサイズが多いほど(基底関数系が大きいほど)、元の形状をより正確に近似できます。
これらの基底関数は、多くの場合、水素原子のような単純な系の解析的な解にヒントを得て、ガウス型やSlater型の関数の形を取りますが、それらもあくまで近似です。理論的には「厳密」でも扱いにくい分子軌道をそのまま扱う代わりに、これらの基底関数の線形結合(係数を持つ和)として表現します。基底関数が原子軌道に似ている場合、この方法は線形結合原子軌道(LCAO)法として知られています。この線形結合の係数を最適化することで、選択した基底関数系の制約の範囲内で最良の近似波動関数とエネルギーを見つけることができます。
- 基底関数系に含まれる関数が多いほど近似の精度は上がりますが、計算コストが高くなります。
- 小さな基底関数系では粗い推定しか得られませんが、大きな基底関数系ではより正確な結果が得られる代わりに、より多くの計算リソースを必要とします。
まとめると、計算を実行可能にしてコストを削減するために、変分原理を用いて波動関数を近似します。これにより計算の複雑さが軽減され、エネルギーを最小化するための反復的な最適化が可能になります。一方、基底関数系のアプローチは、連続的な波動関数を直接解くのではなく、原子軌道をあらかじめ定義された関数の組み合わせとして表現することで計算を単純化します。
理解度チェック
試行波動関数を考えます。ここでは規格化定数、は調整可能なパラメーターです。
(a) 次の条件を満たすを求めることで、試行波動関数を規格化してください:.
Answer
与えられた試行波動関数を規格化するには:
ガウス積分を使用します:
と設定すると:
(b) 次のハミルトニアンの期待値を計算してください:ただしは単純調和振動子ポテンシャルに対応します。
Answer
調和振動子のハミルトニアンは:
運動エネルギーの期待値
2階微分を求めると:
よって:
標準的なガウス積分の結果を用いると:
ポテンシャルエネルギーの期待値
以下を利用します:
すると:
全エネルギーの期待値
(c) 変分原理を用いてを最小化することで最適なを求めてください。
Answer
エネルギーが最小になるようを最適化します。
微分
解くと:
をに代入すると:
これは量子調和振動子の基底状態エネルギーの厳密な値と一致します。
VQE(変分量子固有値ソルバー)
変分量子固有値ソルバー(VQE)は、のプロセスを探求するために使用する主要な手法です。ここではVQEとは何か、どのように機能するかを見ていきます。ただし、その前にまず確認問題を通じて非常に重要な点を確認しましょう。
理解度チェック
化学問題に対してすでにこれほど多くの戦略があるのに、なぜ量子コンピューターが必要なのでしょうか?また、量子コンピューターと古典コンピューターを組み合わせて使用する目的は何でしょうか?
Answer
量子計算は、量子状態の指数的なスケーリングによって古典コンピューターが苦手とする問題に取り組むことで、化学に革命をもたらす可能性があります。リチャード・ファインマンは、自然をシミュレートするには計算もまた量子的でなければならないと有名な言葉を残しています [参考文献1]。
たとえば、最もシンプルな基底関数系(STO-3G)でカフェインをシミュレートするにはビットが必要で、これは観測可能な宇宙における星の総数()をはるかに上回ります [参考文献2]。量子コンピューターは160量子ビットでカフェインの電子軌道を記述できます。
量子コンピューターは、重ね合わせとエンタングルメントを用いることで量子的な相互作用を自然に処理でき、正確な分子シミュレーションを実現する有望な方法を提供します。さらに、量子コンピューター(電子シミュレーション)と古典コンピューター(データの前処理・後処理、アルゴリズムプロセス管理、最適化など)の両方の利点を組み合わせることができます。これらの組み合わせは、材料探索、創薬、反応予測を促進し、コストのかかる試行錯誤の実験を減らすことが期待されています。[参考文献3][参考文献4]
化学問題に量子コンピューターが必要な理由と、量子・古典コンピューティングリソースを両方使用する理由については、以下の記事を参照してください:
それではVQEに戻りましょう。
VQEは量子コンピューターと古典コンピューターの力を組み合わせ、変分原理を基本として系の基底状態エネルギーを求めます。VQEを理解するために、まず3つの部分に分けて考えましょう:
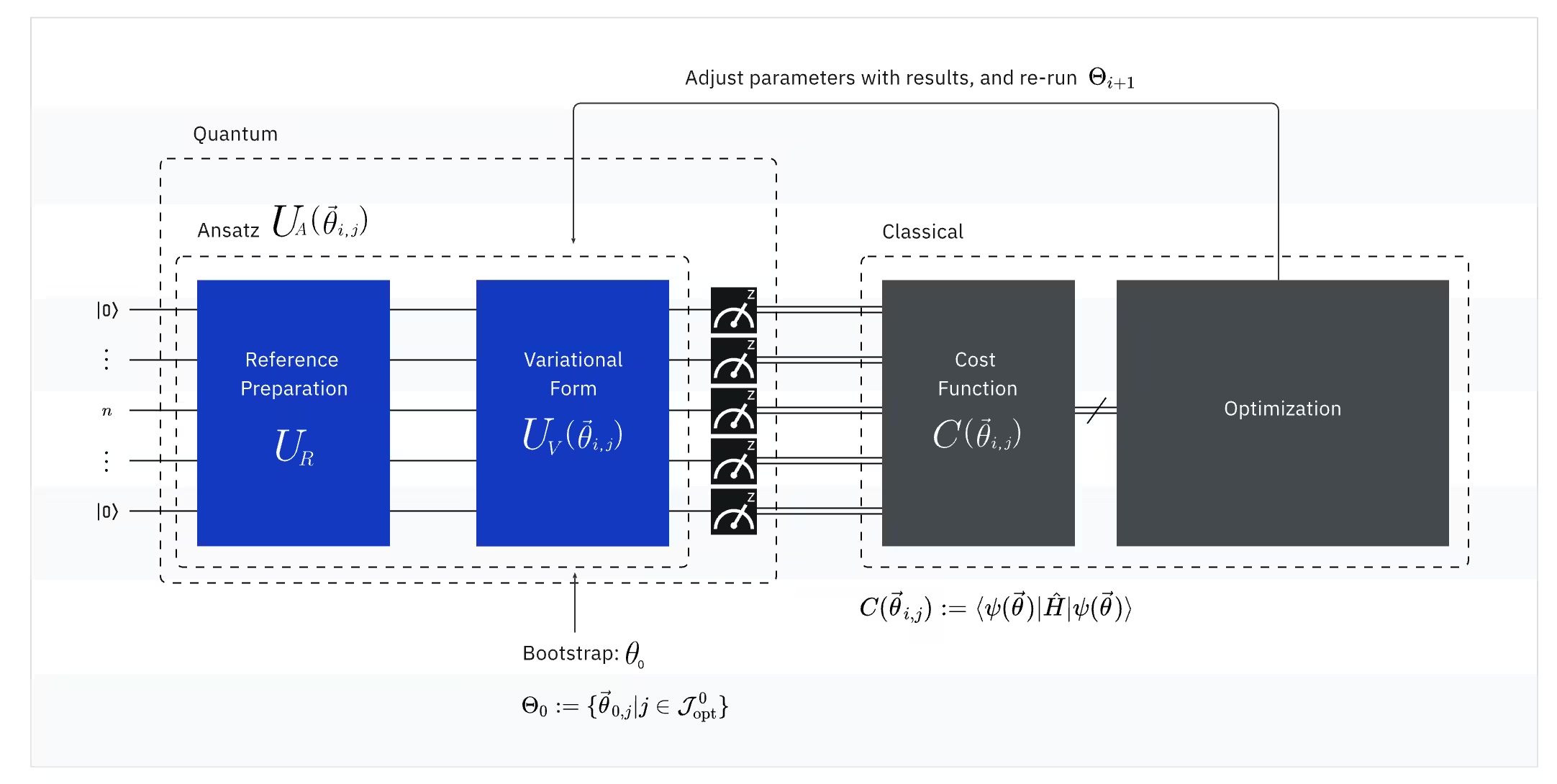
(量子)オブザーバブル:分子ハミルトニアン(分子のエネルギー)
VQEにおいて、分子・原子のハミルトニアンはオブザーバブルです。つまり、実験を通じてその値を測定できます。私たちの目標は、分子の最低エネルギー(基底状態エネルギー)を見つけることです。そのために、パラメーター付き量子Circuit(ansatz)によって生成された試行量子状態を使用します。オブザーバブルを測定し、最低エネルギーに達するまで量子状態を最適化します。
分子ハミルトニアンに使用する基底関数系は、必要なQubitの数を決定し、VQEの精度に直接影響します。適切な基底関数系を選択することは、効率と精度のバランスを保つうえで非常に重要です。基底関数系を変えずに計算を簡略化するために、対称性の活用とアクティブスペース削減などの戦略を使用できます。多くの分子は対称的な形状(蝶や雪の結晶のような)を持ち、一部が同じように振る舞うことを意味します。すべてを個別に計算する代わりに、ユニークな部分だけに注目することで量子リソースを節約でき、対称性を活用することができます。アクティブスペース削減では、すべての電子が分子エネルギーに大きく影響するわけではないため、重要な軌道だけを考慮します。原子核の近くにある電子はほぼ変化しませんが、それ以外の電子は結合に影響を与えます。これらの手法を適用することで、精度を維持しながらVQEをより効率的にすることができます。
適切な基底関数系と上記の戦略を用いて分子ハミルトニアンを得たら、このハミルトニアンを量子コンピューターに適した形に変換する必要があります。問題をパウリ演算子にマッピングすることは非常に複雑になる場合があります。これは特に、Qubitが区別可能であるのに対して区別不可能な粒子(電子)を扱う量子化学において当てはまります。ここではマッピングの詳細には触れませんが、関連するリソースをご紹介します。問題を量子演算子にマッピングする一般的な議論は、実践的な量子計算で見つけることができます。化学問題を量子演算子にマッピングする詳細な議論は、VQEによる量子化学に掲載されています。
このモジュールでは、量子コンピューターの使用に集中できるよう、とに対する適切な(1量子ビット)ハミルトニアンを提供します。これらの1量子ビットハミルトニアンは、STO-6G基底関数系とジョルダン-ウィグナー変換を使用して準備されています。ジョルダン-ウィグナー変換は、1つのスピン軌道の占有を1つのQubitの占有にマッピングするため、最も分かりやすい物理的解釈を持つ最も直接的なマッピングです。また、ハミルトニアンの対称性を利用したQubit削減手法を使用しており、これはスピン占有の振る舞いのパターンを活用してQubitの数を削減します。分子については、2つの水素原子間の距離が0.735 であると仮定します。
(量子)Ansatz:試行波動関数(量子Circuitで試行量子状態を構築する方法)
VQEにおけるansatz(複数形:ansätze)は、2つの主要な要素で構成されています。最初の要素は初期状態準備であり、変分パラメーターを持たない量子Gateを適用してQubitの状態を設定します。2番目の要素はパラメーター付き量子Circuitであり、ラジオのダイヤルのように調整可能なパラメーターを持つ特殊な量子Circuitです。これらのパラメーターは最後の部分である古典的オプティマイザーが最良の基底状態に到達するために使用されます。
変分原理のセクションで学んだように、試行状態の質が変分アルゴリズムの結果の質に影響します。これは、VQEにおいて良いansatzを選ぶことが重要であることを意味します。改めて、これは豊かで複雑なテーマです。ここでは、ansatzの種類やその起源については触れません。パラメーター付き量子CircuitやansatzについてさらにDOQUMENTATIONしたい場合は、変分アルゴリズム設計コースのAnsatzと変分形式レッスンを参照してください。ansätzeの詳細な説明と例が掲載されています。
このモジュールでは1量子ビットハミルトニアンを使用するため、ansatzとして1量子ビットのパラメーター付き量子Circuitが必要です。次のセクションでは3種類の1量子ビットansätzeを見ていきます。それらを比較し、ansatzを選択する際の重要な考慮点について議論します。
(古典)オプティマイザー:量子Circuitの微調整
量子コンピューターがansatzからオブザーバブルのエネルギーを測定すると、ansatzのパラメーターとエネルギー値が古典的オプティマイザーに送られ、調整が行われます。この最適化プロセスは古典コンピューター上で実行され、通常はSciPyのような汎用的な科学計算パッケージを使用します。
古典的オプティマイザーは測定されたエネルギーをコスト関数として扱います。最適化問題において、コスト関数(目的関数とも呼ばれます)は特定の解がどれだけ「良い」かを測定する数学的な関数です。オプティマイザーの目標は、このコスト関数を最小化するパラメーターのセットを見つけることです。分子の基底状態エネルギーを求める文脈では、エネルギー自体がコスト関数として機能します。量子Circuit(私たちの「解」)に対して最低エネルギーを与えるパラメーターを見つけたいのです。古典的オプティマイザーはこの測定されたエネルギー値(コスト)を使用し、量子ansatzの次の最適化されたパラメーターセットを決定します。この更新されたパラメーターが量子Circuitに送り返され、プロセスが繰り返されます。各反復で、古典的オプティマイザーはエネルギーを下げようとパラメーターを調整し(コスト関数を最小化し)、事前に定義された収束基準が満たされるまで続きます。理想的には、(その結合距離と基底関数系での)分子の基底状態に対応する最低エネルギーが見つかります。
SciPyのような科学計算パッケージには多くの最適化戦略が提供されています。詳細は変分アルゴリズム設計コースの最適化ループレッスンを参照してください。ここではCOBYLA(線形近似による制約付き最適化)を使用します。COBYLAは複雑なエネルギーランドスケープに適した最適化アルゴリズムです。特にCOBYLAは、研究対象の関数の勾配を計算しようとしません。これは勾配フリーオプティマイザーと呼ばれます。目を閉じた状態で山岳地帯の最高峰を見つけようとしていると想像してください。全体の地形が見えないため、様々な方向に小さな歩みを進めながら、上がっているか下がっているかを確認します。COBYLAも同様に、パラメーター空間を移動しながら異なる値をテストし、最良の結果が見つかるまで徐々に改善していきます。
これでVQEの計算を実行する準備が整いました。そのために、VQEプロセス全体をまとめた以下の確認問題に取り組んでみてください。
理解度チェック
空欄に正しい用語を入れて、VQEプロセスの概要を完成させてください。答えを確認するにはクリックしてください。
VQEは変分量子アルゴリズムであり、(1) ________と古典コンピューティングの力を組み合わせて分子の(2) __________を求めるために使用されます。プロセスは(3) __________を定義することから始まります。これは系の全エネルギーを表し、量子測定におけるオブザーバブルとして機能します。次に(4) __________を準備します。これは分子の試行波動関数を表す調整可能なパラメーターを持つ量子Circuitです。これらのパラメーターは(5) __________を使用して最適化されます。これは測定されたエネルギーを最小化するためにパラメーターを反復的に調整する古典アルゴリズムです。上記の議論では(6) __________オプティマイザーを使用しており、これは微分計算を必要とせずにansatzパラメーターを調整します。プロセスは(7) __________に達するまで続き、分子の最低エネルギーが見つかったことを意味します。
ワードバンク:
- 古典的オプティマイザー
- 基底状態エネルギー
- ハードウェア効率的
- ansatz
- 分子ハミルトニアン
- COBYLA
- 量子コンピューティング
- 収束
Answer
1 → 量子コンピューティング
2 → 基底状態エネルギー
3 → 分子ハミルトニアン
4 → ansatz
5 → 古典的オプティマイザー
6 → COBYLA
7 → 収束
VQEを用いた水素原子の基底状態エネルギーの計算
それでは、学んだことを活用して水素原子の基底状態エネルギーを計算しましょう。モジュール全体を通じて、「Qiskitパターン」と呼ばれる量子計算のフレームワークを使用します。このフレームワークはワークフローを以下のステップに分解します:
- ステップ1:古典的な入力を量子問題にマッピングする
- ステップ2:量子実行のために問題を最適化する
- ステップ3:Qiskit Runtimeプリミティブを使用して実行する
- ステップ4:後処理と古典的分析

基本的にこれらのステップに従います。
まず、Qiskit Runtimeプリミティブを含む必要なパッケージを読み込みましょう。また、利用可能な最も空いている量子コンピューターを選択します。
以下に初回使用時に認証情報を保存するためのコードがあります。保存後はノートブックからこの情報を必ず削除してください。ノートブックを共有する際に認証情報が誤って共有されないようにするためです。詳細については、IBMクラウドアカウントの設定と信頼できない環境でのサービスの初期化を参照してください。
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
以下のセルを使用すると、ノートブック全体でシミュレーターと実際のハードウェアを切り替えることができます。今すぐ実行することをお勧めします:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
ステップ1:問題を量子回路と演算子にマッピングする
まず、特定の結合距離における水素分子()のハミルトニアンを定義することでVQEの計算を開始します。このハミルトニアンは、標準的な手順に従って分子系から生成・マッピングされた結果として、量子ビット演算子の形でシステムの全エネルギーを表します。具体的には、1) STO-6G基底関数系(電子軌道を近似するための数学的関数の集合)を用い、2) ジョルダン–ウィグナー変換(電子を記述するフェルミオン演算子を量子ビット演算子に変換する手法)を適用し、3) ハミルトニアンのパリティを利用した量子ビット削減によって問題を簡略化する、という手順を踏んでいます。
前述のとおり、計算された基底状態エネルギーは基底関数系の選択や分子の幾何構造(結合距離など)に大きく依存します。この特定の構成と上記の変換を経た結果、得られる量子ビットハミルトニアンは次のようにシンプルな形になります。
ここで、 は恒等演算子、 はパウリ Z 演算子を表し、いずれも単一の量子ビットに作用します。係数は、この特定の結合距離における STO-6G 基底関数系を用いて計算された積分と適切な変換から導かれます。
このハミルトニアンが定義できたので、VQE を用いてその基底状態エネルギーを計算することができます。計算された基底状態エネルギーを期待値と比較することは有益です。単一の孤立した水素原子(H)の基底状態エネルギーは、(相対論的効果を無視した場合)ちょうど −0.5 ハートリーです。ここで、上記で定義したこの特定の量子ビットハミルトニアンの厳密な基底状態エネルギーを計算し、関連する既知の値と比較してみましょう。
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
次に、基底状態の試行波動関数 を準備するために、パラメータ化された量子回路、すなわちアンザッツが必要です。目標は、エネルギー期待値 を最小化するパラメータ を見つけることです。アンザッツの選択は非常に重要です。なぜなら、Circuit が準備できる量子状態の集合を決定するからです。「良い」アンザッツとは、調べているハミルトニアンの真の基底状態に非常に近い状態を表現できるほど柔軟でありながら、現在の量子コンピュータには多すぎるパラメータや深すぎる Circuit を必要としない程度の複雑さに留まっているものです。
ここでは、1つの量子ビットで構成された3種類の異なるアンザッツを試し、どれが単一量子ビットの取りうる量子状態をより広く「カバー」できるかを確認します。「カバレッジ」とは、アンザッツ Circuit がパラメータを変化させることで生成できる量子状態の範囲を指します。
単一量子ビット回転 Gate の異なる組み合わせに基づく3つのアンザッツを使用します。
- 1軸回転 Gate アンザッツ:このアンザッツは単一の軸()周りの回転のみを使用します。ブロッホ球上では、特定の円に沿った移動のみに対応します。最も柔軟性が低く、限られた状態の集合しかカバーできません。
- 2軸回転 Gate アンザッツ(2種類):これらのアンザッツは2つの異なる軸周りの回転を組み合わせます( および )。単一軸の回転と比較して、ブロッホ球のより広い領域に到達することができます。
これら3つのアンザッツで得られた VQE の結果を比較することで、アンザッツの柔軟性と状態空間のカバレッジが、簡略化されたハミルトニアンの真の基底状態エネルギーを見つける能力にどのような影響を与えるかを確認できます。より柔軟なアンザッツはより良い近似を見つける可能性を持ちますが、古典的なオプティマイザにとってより難しくなる場合もあります。
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
次に、各パラメータに対して5000個の乱数を生成し、これら3つのアンザッツがランダムなパラメータで生成したランダムな量子状態の分布をプロットしてみましょう。これらのパラメータは、球面上の異なる軸周りの回転角のようなものと考えることができます。量子状態の分布を確認するために、ブロッホ球を使用します。これは単一量子ビットの状態を示す3次元の球体です。球上の任意の点は量子ビットの取りうる状態を表しており、北極と南極は古典的な「0」と「1」に対応しますが、量子ビットはその中間のどの位置にも存在でき、重ね合わせのような特別な量子特性を示します。まず、3次元のブロッホ球をプロットするための必要な関数を準備し、5000個のランダムなパラメータを用意します。
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
最初のアンザッツがどのように機能するか見てみましょう。
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
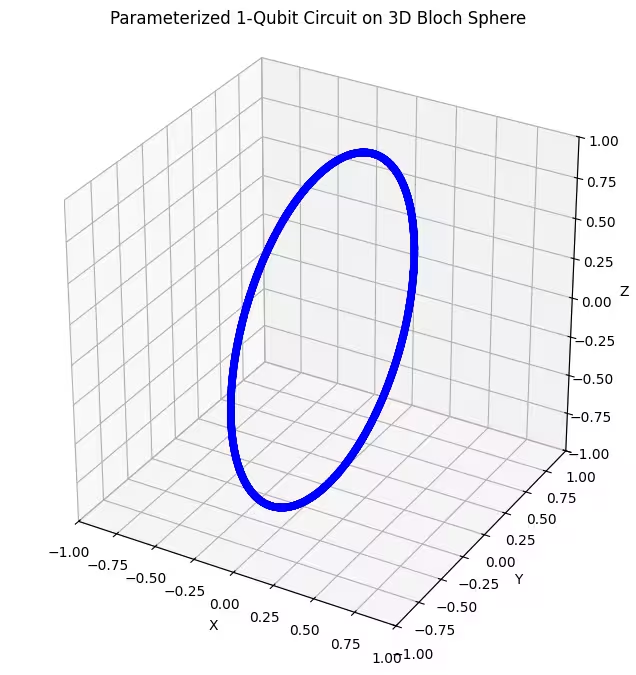
最初のアンザッツはブロッホ球上でリング状に分布した量子状態を返すことがわかります。これは、アンザッツに単一の回転パラメータのみを与えているためで、1つの軸周りに回転した状態しか生成できません。点 を出発点として1つの軸周りに回転すると、常にリングが得られます。次に、直交する2つの回転 Gate(Rx と Rz)を持つ2番目のアンザッツを確認しましょう。
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
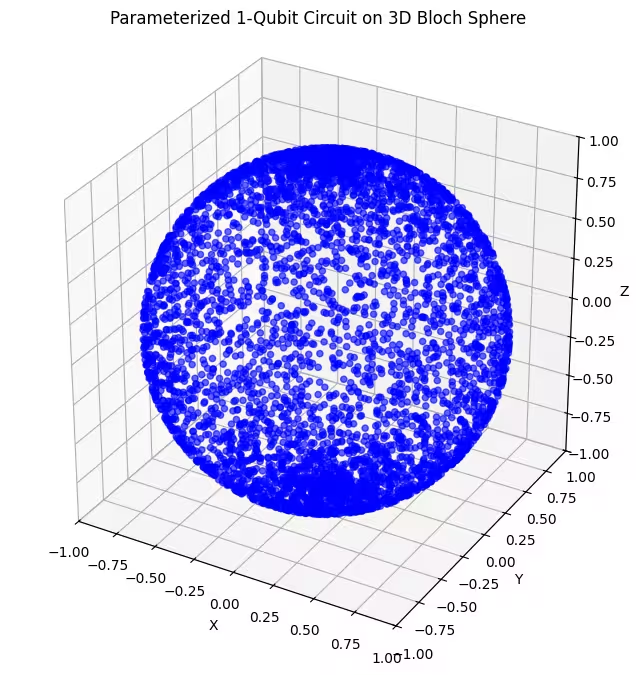
2番目のアンザッツはブロッホ球のより広い領域をカバーしていますが、点が極周辺に集中しており、赤道付近では広がっていることに注目してください。それでは最後のアンザッツを確認しましょう。
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
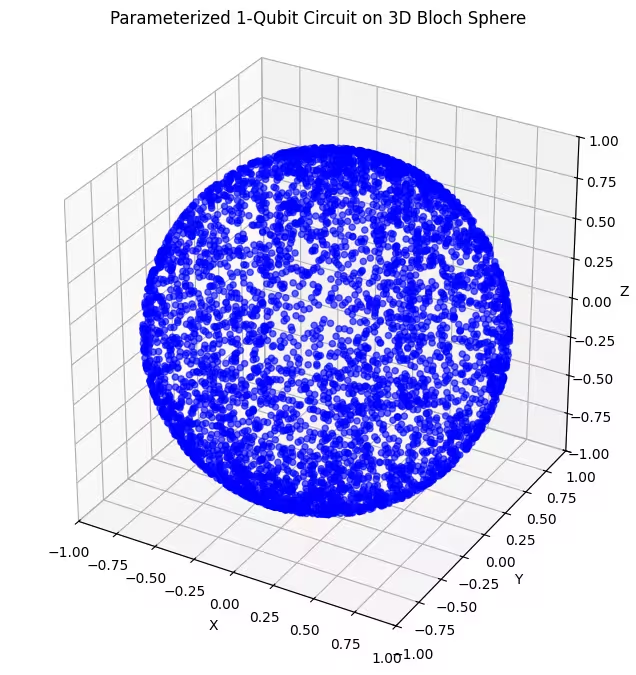
最後のアンザッツによって生成された量子状態がより均一に分布していることがわかります。
先述のとおり、最善のアプローチは探している基底状態についての知識を深め、その基底状態に近い状態を調べるのに適したアンザッツを選択することです。たとえば、基底状態が極付近にあるとわかっている場合は、アンザッツ2を選択するかもしれません。ここでは簡単のために、ブロッホ球全体を均一に探索するアンザッツ3を使用します。
アンザッツが選択できたので、Circuit を描画してみましょう。
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

ステップ2:対象ハードウェアに向けて最適化する
実際の量子コンピュータで計算を実行する場合、量子 Circuit のロジックだけを考慮すれば良いわけではありません。特定の量子コンピュータが実行できる演算の種類や、使用する qubit が量子コンピュータ上のどこに配置されているかも重要です。それらは隣接していますか?それとも離れていますか?そのため、次のステップは、使用する量子コンピュータにとってネイティブな Gate を用いて Circuit を書き直し、qubit のレイアウトを考慮することです。これは transpilation(トランスパイル)によって実現できます。このプロセスの後、単純なアンザッツが別の Gate のセットに変換され、抽象的な qubit が実際の量子コンピュータ上の物理的な qubit にマッピングされる様子を確認できます。
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

アンザッツの rx, rz Gate が、Backend のネイティブ Gate である rz, sx Gate の系列に変換されていることがわかります。また、q0 が5番目の物理 qubit にマッピングされていることも確認できます。さらに、これらの変更に応じてハミルトニアンもマッピングする必要があります。以下のコードで行います。
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
ステップ3:対象ハードウェアで実行する
いよいよ実際の QPU で VQE を実行する時です。そのためには、まず最適化プロセスのためのコスト関数が必要です。この関数は、アンザッツによって生成された量子状態でハミルトニアンの期待値を評価します。ご心配なく!すべてを自分でコーディングする必要はありません。この関数を用意しましたので、以下のセルを実行するだけです。
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
最後に、アンザッツの初期パラメータとその最適化プロセスを準備します。すべてゼロまたは乱数を使用することができます。以下のセルでは初期パラメータを選択していますが、0 から の一様分布でランダムにサンプリングする行をコメント・アンコメントして自由に試してみてください。
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
おめでとうございます!初めての量子化学実験を無事に完了しました。ハミルトニアンの厳密な基底状態エネルギーと今回の結果の間に差異は見られますが、デフォルトのエラー緩和技術(読み出しエラーを補正する)を使用したため、差は小さいものとなっています。非常に良いスタートです!
注:resilience_level を使用してエラー緩和のレベルを設定することで、より良い結果を得ることができます。デフォルト値は1であり、より高い値を設定すると QPU の使用時間は増加しますが、より良い結果が返ってくる可能性があります。
ステップ4:後処理
古典的なオプティマイザーがどのように機能したかを確認してみましょう。以下のセルを実行して、収束のパターンを確認してください。
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

初期値がかなり良好な値から始まったため、わずか10ステップで良好な最終値が得られました。大小さまざまなピークが見られますが、これはCOBYLAオプティマイザーの典型的な特徴です。このオプティマイザーはエネルギー地形を直接「見る」ことなく空間を探索し、各測定ごとにステップサイズを調整します。
理解度確認
どのような観察ができましたか?理論値に近い結果、またはハミルトニアンの正確な基底状態エネルギーに近い結果を得るために、上記のプロセスのどの部分を改善できるでしょうか?また、その際に考慮すべき事項は何でしょうか?
Answer
まず考慮すべきは、分子のハミルトニアン計算に使用する基底関数セットの変更です。先述のとおり、H原子の基底状態エネルギーは-0.5 Hartreeであることがよく知られています。しかし、ここで選択したSTO-6G基底では、この値を正確に導出するには不十分です。
より複雑な基底を選択すると、ハミルトニアンが使用するQubitの数が増加するため、量子化学問題に適したより複雑なansatzを選ぶ必要があります。
次に最適化すべきは、QPUにおけるノイズの管理です。より高度なエラー緩和技術を用いると、より良い結果が得られますが、処理に時間がかかる場合があります。また、shot_numberが結果に与える影響についても検討してください。
最後に、異なるオプティマイザーを試すことで、より良い収束性能を達成できる可能性もあります。
VQEによる水素分子の基底状態エネルギーの計算
原子を使ったVQEの全体的なプロセスを確認したので、今度は分子の基底状態エネルギーをより迅速に計算します。
ステップ1:問題を量子Circuit・演算子にマッピングする
ここでも、STO-6G基底とJordan-Wigner変換を使用し、ハミルトニアンのパリティを利用したQubit削減を適用した1-QubitのハミルトニアンをおいてAnを提供します。2つの水素原子間の原子間距離として0.735 を使用しています。
単一の水素原子()の計算とは異なり、水素分子()の基底状態を計算するには、電子軌道に関連するエネルギーに加えて、2つの水素原子の核間に働く反発力も考慮する必要があります。このステップでは、この値を定数として与えます。この値の実際の計算はチェックイン問題で行います。
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
ステップ2:ターゲットハードウェア向けに最適化する
以前のVQEとハミルトニアンが使用するQubit数は、実行に使用するBackendと同じであるため、既存のansatzとその最適化済みの形式をそのまま使用します。
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
ステップ3:ターゲットハードウェアで実行する
いよいよ実際のQPUで計算を行います。ほぼすべての設定は同じですが、ハミルトニアンに合わせた適切な初期点を使用します。また、反復処理の部分では、QPU上でansatzに対するハミルトニアンの期待値を計算するために使用するEstimatorの設定が、以前の計算と若干異なります。この変更については、チェックイン問題でさらに詳しく説明します。
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
VQEは理論上、真の基底状態エネルギーの上限を与えるとされていますが、実際の量子ハードウェアやノイズのある量子シミュレーター上での実装では、基底関数セットやQubit削減といったハミルトニアン準備における近似が誤差を生じさせ、測定されたエネルギーが厳密な理論値や特定の数値的な参照値をわずかに下回る場合があります。多少の誤差はありますが、特に反復回数が少ないことを考慮すると、結果は満足のいくものと言えます。それでは、オプティマイザーの動作を確認して、このVQE計算を締めくくりましょう。
ステップ4:後処理
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

理解度確認
定数値(0.71997 Hartree)として含めた分子の核間反発エネルギーを計算してみましょう。

クーロンの法則と原子単位系を用いて値をHartree単位で求めてください。
Answer
両方の水素核は正に帯電しているため、静電気力によって互いに反発します。この反発はクーロンの法則で記述されます:
ここでは陽子の電荷、は真空の誘電率、はメートルまたはボーア半径で測った2つの核間の距離(単位はジュール(J))です。
このエネルギーをHartreeで計算するには、上記の式を原子単位系(AU)に変換する必要があります。AUでは、、ボーア半径()は1であり、AUにおける基本的な長さの単位となります。これらの簡略化により、クーロンの法則は次のように簡約されます:
ここではボーア半径()で表す必要があります。
で与えられた核間距離をに変換するには、以下の変換式を使用します:
したがってはとなります。
したがって、与えられたの核間反発エネルギーは
の反応エネルギーを計算する
ここまでで得た結果を活用しましょう!変分量子固有値ソルバーであるVQEを使用して、原子と分子の基底状態エネルギーを計算しました。残るは、得られた値を使ってのプロセスの反応エネルギーを求めることです。
反応エネルギーとは、物質が反応して新しい物質を生成する際のエネルギー変化です。何かを組み立てるときをイメージしてください:エネルギーを加える必要がある場合(ブロックを積み上げるようなもの)もあれば、エネルギーが放出される場合(ボールが坂を転がり下りるようなもの)もあります。化学では、反応はエネルギーを吸収する(吸熱反応)か、エネルギーを放出する(発熱反応)かのどちらかです。
プロセスの反応エネルギーは、以下の式で計算できます:
以下のセルを実行して、この結果を視覚的に確認しましょう。ここでは各ハミルトニアンの厳密な基底状態値を使用し、厳密解とVQEの結果による反応エネルギーを比較します。
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

図に示すように、多少の誤差はあるものの、ハミルトニアンの厳密な基底状態エネルギーとVQEの結果を使って計算した反応エネルギーはよく一致しており、どちらも-0.2 Hartree付近の値となっています。
ここで注目すべき点は、このプロセスの反応エネルギーが負の値であることです。これは、このプロセスを通じてエネルギーが放出されること、すなわち生成された分子は2つの単独原子よりもエネルギーが低いことを意味します。
- まとめ
ここまで学んだ内容を整理しましょう。
まず、量子化学問題を解くために必要な2つの重要な近似技術として、変分原理と基底関数セットの選択を学びました。どちらもVQEの基礎となる概念です。変分原理については、単純調和振動子の基底状態エネルギーを手計算で求めることで確認しました。
次に、量子系の基底状態エネルギーを計算するために広く使われているアルゴリズムであるVQEを探求しました。実際にコードを実行して、水素原子()と水素分子()の基底状態エネルギーを計算しました。特に、対象系に適切な分子ハミルトニアンを取得し、それを量子コンピューター上で実行可能な形式に変換する必要があることを学びました。また、ansatz(パラメータ化された量子Circuit)がVQE内で試行量子状態を準備するために必要であることを理解し、適切なansatz Circuit構造を選択することの重要性について議論しました。さらに、VQEが古典コンピューターを使った反復最適化プロセスに依存して量子Circuitを最低エネルギー状態へと導くことを学び、そのプロセスがどのように収束するかを確認しました。
最後に、VQEを通じて得られたとの基底状態エネルギーを使用して、のプロセスの反応エネルギーを計算しました。
VQEは強力なニアタームの量子アルゴリズムですが、その限界についても認識しておくことが重要です。VQEの性能はansatzの選択に大きく依存します。より大きく複雑な分子に対して、真の基底状態を正確に表現できる効率的に準備可能なansatzを見つけることは困難になります。さらに、現在の量子ハードウェアはノイズの影響を受けやすく、特に深いCircuitやより多くのQubitを使用する場合、VQEの結果の精度に影響を与えることがあります。これらの課題にもかかわらず、VQEは基礎的なアルゴリズムとして機能しており、ニアタームの量子コンピューターで量子化学においてできることの限界を拡大するために、より高度な変分手法やエラー緩和技術の研究が続けられています。たとえば、サンプルベース量子対角化(SQD)のようなアルゴリズムが開発されつつあります。これは、量子Circuitから得られたサンプルと、部分空間における古典的な対角化を組み合わせることで、エネルギー推定の精度を向上させ、VQEが直面する特に測定効率とノイズ耐性に関する課題のいくつかに対処するアプローチです。
確認と問題
重要な概念
- 変分量子アルゴリズムは、古典コンピューターと量子コンピューターが協調して問題を解く計算パラダイムです。
- VQEでは、まず対象系のハミルトニアンを量子コンピューター上で実行できるようにQubitにマッピングします。次にパラメーター化された量子Circuit(アンザッツ)を選択し、アンザッツのパラメーターを変化させながら繰り返し測定を行い、最小エネルギー値に達するまで最適化を続けます。パラメーター空間の探索は古典的オプティマイザーを用いて行います。良好な結果を得るためには、適切なアンザッツと適切なオプティマイザーを選択することが重要です。
- 反応エネルギーとは、化学反応における全エネルギー変化であり、反応物と生成物のエネルギーの差によって決まります。
正誤問題
- 変分原理とは、任意の試行波動関数に対するエネルギーの期待値は、常に真の基底状態エネルギー以上であると述べるものである。
- 基底関数系(basis set)とは、量子波動関数を近似するために使用される関数の集合である。
- VQEは、与えられたハミルトニアンに対してシュレーディンガー方程式を厳密に解くための量子アルゴリズムである。
- VQEでは、パラメーター化された量子Circuit(アンザッツ)を用いて試行波動関数を準備する。
- VQEにおけるオプティマイザーの選択(例:COBYLA、SPSA、ADAM)は結果の品質に影響しない。
- Qiskitの
EstimatorはVQEにおいてハミルトニアンの期待値を直接計算するために使用される。
多肢選択問題
- VQEにおけるハミルトニアンの役割は何ですか?
- A) ランダムな量子状態を生成する
- B) 量子状態のエネルギーを決定する
- C) 量子Circuitを最適化する
- D) エンタングルメントを生成する
- VQEアルゴリズムの主な目的は何ですか?
- A) ハミルトニアンの基底状態エネルギーを求める
- B) Qubit間のエンタングルメントを生成する
- C) グローバーの探索を実行する
- D) RSA暗号を解読する
- このノートブックでアンザッツを比較するために生成される量子状態の数はいくつですか?
- A) 100
- B) 1000
- C) 5000
- D) 10,000
- VQEに古典的オプティマイザーが必要な理由は何ですか?
- A) 量子測定を実行するため
- B) アンザッツのパラメーターを更新してエネルギーを最小化するため
- C) Qubitをエンタングルさせるため
- D) 量子ランダム性を生成するため
- アンザッツがパラメーター化されて設計される理由は何ですか?
- A) 量子状態の準備を可能にするため
- B) 幅広い量子状態空間を探索できるようにするため
- C) Circuitの複雑さを減らすため
- D) 固有値を直接測定するため
- 良いアンザッツを選ぶことについて、最も正確な記述はどれですか?
- A) アンザッツはブロッホ球上に均等に分布した状態を生成しなければならず、そうでなければ失敗する。
- B) アンザッツは対象系に合わせて調整し、基底状態に近い状態を生成できるようにすべきである。
- C) アンザッツは変分パラメーターを使ってランダムな状態を生成すべきである。
- D) より良いアンザッツは常により多くの変分パラメーターを持つ。
(オプション)付録:アンザッツの複雑さによるオプティマイザーのオーバーヘッド
VQEにはいくつかのよく知られた課題[ref 6]があり、以下はこれまでに学んだ内容と関連するものです。
- アンザッツ選択の課題
適切な変分アンザッツを選択することには本質的な難しさがあります。化学にインスパイアされたアンザッツ(UCCSDなど)は物理的な精度を提供しますが、深いCircuitを必要とします。一方、ハードウェア効率の良いアンザッツはCircuitが浅いですが、物理的な解釈可能性に欠ける場合があります。また、多くのアンザッツは過剰な変分パラメーターを導入しており、精度の向上にはほとんど貢献しないにもかかわらず、最適化の難しさを大幅に増大させます。
- 最適化の困難さ
VQEの最適化ランドスケープには、勾配が指数関数的に消滅する領域(バレン・プラトー)が存在する場合があり、古典的オプティマイザーが変分パラメーターを効率よく更新することが難しくなります。この問題に対して、研究者たちは勾配あり・勾配なしのさまざまなオプティマイザーを試みてきましたが、いずれも課題を抱えています。勾配ベースのオプティマイザーはバレン・プラトーに悩まされ、勾配なしの手法は多数の関数評価を必要とします。
- オプティマイザーのオーバーヘッド
もう一つのよく知られた課題がオプティマイザーのオーバーヘッドであり、これは問題の規模に関連しています。VQEに必要な量子Circuitは、問題のサイズが大きくなるにつれて深さと複雑さが増します。これに伴い、最適化すべきパラメーターの数も通常増加します。パラメーター数の増加に伴い最適化プロセスが扱いにくくなり、収束が遅くなるとともに最適解の探索が困難になります。
ここでは、 分子に対してVQEを使用し、2種類のアンザッツを用いてこれらの課題を確認します。
(注意:これはQPU時間を多く消費する可能性があります。時間が十分でない場合は、シミュレーターを使用しても構いません。)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
次に、全て1で初期化した初期点を使用し、最大20ステップのVQEを実行して、2つの実行の収束を比較します。
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

上のグラフは、変数の多いアンザッツの最適化プロセスが安定した収束に達するまでにより多くの時間を要することを明確に示しています。
単純な単一QubitのCircuitや素直なアンザッツに頼るのではなく、より大きな量子Circuitや複雑な構造を持つアンザッツが必要になると最適化の複雑さが増します。これはVQEにおけるよく知られた課題、すなわちオプティマイザーのオーバーヘッドを浮き彫りにしています。
研究者たちは、量子コンピューターを化学問題に活用するためのさまざまな高度な方法論を引き続き開発しています。IBM Quantum Learningでは、多様な教育資料にアクセスできます。
参考文献
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)